- SOLUTION (ÉQUILIBRES EN)
- SOLUTION (ÉQUILIBRES EN)Dans de nombreux domaines de la chimie, les réactions ont lieu en solution. Citons les grandes préparations de la chimie inorganique, l’hydrométallurgie où l’on met en œuvre l’attaque des minerais par des solutions acides ou basiques, la biochimie, le nucléaire avec le retraitement des combustibles, l’analyse chimique avec les titrages volumétriques et coulométriques. On peut y adjoindre certaines réactions de la chimie organique.La notion d’équilibre chimique tient une large place en chimie des solutions. Les réactions mises en jeu sont régies par les règles classiques découlant de la loi d’action de masses, pour autant qu’elles soient rapides. C’est en général le cas si on excepte bon nombre de réactions d’oxydoréduction et certaines réactions hétérogènes où il faut tenir compte de la vitesse de formation et d’évolution des précipités.L’état d’une solution est en principe parfaitement défini par la connaissance des quantités introduites (les bilans-matière) et du modèle qui régissent l’ensemble des équilibres mis en jeu, chacun de ces équilibres se traduisant par une constante d’équilibre. Moyennant diverses simplifications, on peut obtenir des solutions approchées qui sont suffisantes pour certaines applications; on peut maintenant, à l’aide des moyens de calcul modernes, résoudre tout problème de chimie des solutions de manière complète et rigoureuse.L’introduction d’un soluté au sein d’un solvant implique l’existence d’interactions variées très énergétiques. L’interaction la plus simple tient son origine dans le caractère polaire des molécules de la plupart des solvants usuels – eau, alcools, amides... (interactions ion-dipôle ou dipôle-dipôle). Pendant la dissolution, le solvant exerce une action de solvatation (hydratation dans le cas de l’eau) au cours de laquelle chaque espèce dissoute (ion ou molécule) s’entoure d’un cortège de molécules de solvant. Il peut également exercer une action de solvolyse au cours de laquelle certaines liaisons sont rompues avec apparition d’espèces nouvelles. Le solvant constitue, en outre, un milieu diélectrique où les forces d’attraction s’exerçant entre ions de signes contraires sont affaiblies.L’eau manifeste ces aptitudes de manière très marquée. Elle dissout un grand nombre de composés. Pour certains, les ions préexistant à l’état solide sont hydratés (cas du chlorure de sodium); pour d’autres, les liaisons sont ionisées (cas du chlorure d’hydrogène). La constante diélectrique très élevée ( 﨎 = 80) permet la séparation des ions hydratés (dissociation ionique ), au point qu’ils peuvent être considérés comme indépendants.Ces deux caractéristiques (pouvoir ionisant et pouvoir dissociant) de chaque solvant expliquent son rôle spécifique vis-à-vis des propriétés chimiques des espèces en solution.Toutes les réactions en solution homogène se ramènent à trois types principaux: les réactions d’oxydoréduction, les réactions acide-base, les réactions de formation de complexes. Lorsqu’un composé a une solubilité limitée, il convient d’y ajouter les réactions de précipitation. Ces quatre types de réactions sont souvent mêlés. Des analogies entre les trois types de réactions en phase homogène apparaissent et, de ce fait, en adoptant la notion de couple donneur-accepteur d’une particule, un seul mode de raisonnement permet de les expliquer.1. Réactions acide-base (transfert de protons)DéfinitionsAcides et basesSelon les concepts introduits par J. N. Brönsted, les acides sont des composés susceptibles de céder des protons : ce sont des donneurs de cette particule. Dans le même temps, les bases , composés à même d’en fixer, en sont des accepteurs . À tout acide correspond une base conjuguée et inversement; par suite, un couple acide-base est défini par la relation:
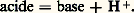 Par exemple, l’anion F size=1漣 est la base conjuguée de l’acide HF:
Par exemple, l’anion F size=1漣 est la base conjuguée de l’acide HF: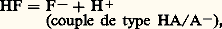 de même, l’ion ammonium NH+4 est l’acide conjugué de la base ammoniac:
de même, l’ion ammonium NH+4 est l’acide conjugué de la base ammoniac: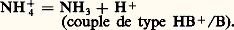 De nombreux composés organiques dont la molécule contient des atomes d’azote, d’oxygène ou de soufre, porteurs de doublets libres (face=F0019 令漣N, 略漣漣O, 略漣漣S), sont des bases; c’est le cas de l’aniline:
De nombreux composés organiques dont la molécule contient des atomes d’azote, d’oxygène ou de soufre, porteurs de doublets libres (face=F0019 令漣N, 略漣漣O, 略漣漣S), sont des bases; c’est le cas de l’aniline: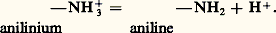 De nombreux cations métalliques se comportent comme des acides en solution aqueuse, par suite de la formation de complexes hydroxyde:
De nombreux cations métalliques se comportent comme des acides en solution aqueuse, par suite de la formation de complexes hydroxyde: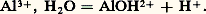 Ce phénomène est connu sous le nom d’hydrolyse .Certaines espèces, comme HC-3, peuvent jouer le rôle d’acide dans un premier système, de base dans un autre système:
Ce phénomène est connu sous le nom d’hydrolyse .Certaines espèces, comme HC-3, peuvent jouer le rôle d’acide dans un premier système, de base dans un autre système: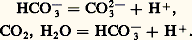 On les appelle des ampholytes . Ce sont des formes intermédiaires entre des polyacides , qui peuvent céder successivement plusieurs protons, et des polybases (dans l’exemple précédent C2, H2O est un diacide, C32- une dibase).Les molécules des solvants peuvent elles-mêmes jouer le rôle d’acides et de bases; ainsi, l’eau joue le rôle de base dans le couple:
On les appelle des ampholytes . Ce sont des formes intermédiaires entre des polyacides , qui peuvent céder successivement plusieurs protons, et des polybases (dans l’exemple précédent C2, H2O est un diacide, C32- une dibase).Les molécules des solvants peuvent elles-mêmes jouer le rôle d’acides et de bases; ainsi, l’eau joue le rôle de base dans le couple: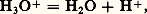 et d’acide dans le couple:
et d’acide dans le couple: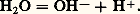 Un solvant pouvant jouer aussi bien le rôle de base (solvant protophile ) que d’acide (solvant protogène ), comme l’eau dans les deux couples précédents, est dit amphiprotique .Réactions acide-baseLe proton n’existant – pratiquement – pas à l’état libre en solution, un acide ne peut jouer son rôle de donneur de cette particule que s’il se trouve en présence d’une base appartenant à un autre couple, à même de la fixer. Entre deux couples acide-base notés 1 et 2:
Un solvant pouvant jouer aussi bien le rôle de base (solvant protophile ) que d’acide (solvant protogène ), comme l’eau dans les deux couples précédents, est dit amphiprotique .Réactions acide-baseLe proton n’existant – pratiquement – pas à l’état libre en solution, un acide ne peut jouer son rôle de donneur de cette particule que s’il se trouve en présence d’une base appartenant à un autre couple, à même de la fixer. Entre deux couples acide-base notés 1 et 2: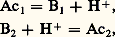 on observera une réaction de la forme:
on observera une réaction de la forme: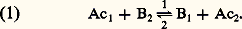 Chaque réaction acide-base apparaît donc comme une réaction d’échange de protons, par exemple:
Chaque réaction acide-base apparaît donc comme une réaction d’échange de protons, par exemple: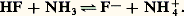 Prévision des réactionsForce des acides et des basesPour chaque réaction acide-base, l’important est d’en connaître le sens prédominant.Si l’équilibre (1) est déplacé dans le sens 1 (réaction de Ac1 sur B2), l’acide 1 est dit plus fort que l’acide 2. Ainsi, plus un acide est fort, plus sa base conjuguée est faible et réciproquement.Il suffit donc, pour permettre une prévision aisée des réactions, d’établir un classement de l’ensemble soit des acides, soit des bases. D’où l’idée, par exemple, dans le cas de la prévision des réactions en solution aqueuse, d’établir un classement des acides selon l’intensité de leur réaction avec l’eau, jouant le rôle de base dans le couple H3+/H2O.Classement des couples acide-baseConstante d’aciditéConsidérons l’acide HA du couple HA/A- (on pourrait raisonner de même dans le cas du couple HB+/B).La réaction d’échange de protons avec l’eau jouant le rôle de base s’écrit:
Prévision des réactionsForce des acides et des basesPour chaque réaction acide-base, l’important est d’en connaître le sens prédominant.Si l’équilibre (1) est déplacé dans le sens 1 (réaction de Ac1 sur B2), l’acide 1 est dit plus fort que l’acide 2. Ainsi, plus un acide est fort, plus sa base conjuguée est faible et réciproquement.Il suffit donc, pour permettre une prévision aisée des réactions, d’établir un classement de l’ensemble soit des acides, soit des bases. D’où l’idée, par exemple, dans le cas de la prévision des réactions en solution aqueuse, d’établir un classement des acides selon l’intensité de leur réaction avec l’eau, jouant le rôle de base dans le couple H3+/H2O.Classement des couples acide-baseConstante d’aciditéConsidérons l’acide HA du couple HA/A- (on pourrait raisonner de même dans le cas du couple HB+/B).La réaction d’échange de protons avec l’eau jouant le rôle de base s’écrit: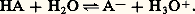 Cet équilibre est caractérisé par la constante:
Cet équilibre est caractérisé par la constante: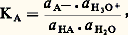 ai désignant l’activité de l’espèce i ; avec les conventions (activité prise égale à 1 pour le solvant) et approximations usuelles (activité confondue avec la concentration pour toutes les espèces présentes diluées en solution), il vient:
ai désignant l’activité de l’espèce i ; avec les conventions (activité prise égale à 1 pour le solvant) et approximations usuelles (activité confondue avec la concentration pour toutes les espèces présentes diluées en solution), il vient: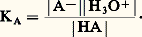 La constante KA ainsi définie est appelée constante d’acidité dans l’eau du couple acide-base considéré. Les constantes KA étant généralement très petites devant 1, on utilise très souvent, à des fins de commodité d’écriture, une échelle logarithmique, introduisant la notation:
La constante KA ainsi définie est appelée constante d’acidité dans l’eau du couple acide-base considéré. Les constantes KA étant généralement très petites devant 1, on utilise très souvent, à des fins de commodité d’écriture, une échelle logarithmique, introduisant la notation: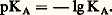 Par exemple, pour l’acide fluorhydrique en solution aqueuse et à 20 0C, les tables de constantes indiquent, pour H- pKA = 3,2, correspondant à:
Par exemple, pour l’acide fluorhydrique en solution aqueuse et à 20 0C, les tables de constantes indiquent, pour H- pKA = 3,2, correspondant à: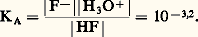 De même, dans le cas du cation ammonium NH4+/NH3, pKA = 9,2, correspondant à:
De même, dans le cas du cation ammonium NH4+/NH3, pKA = 9,2, correspondant à: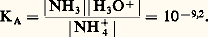 La réaction de transfert de protons entre l’eau jouant le rôle d’acide et elle-même jouant le rôle de base suivant:
La réaction de transfert de protons entre l’eau jouant le rôle d’acide et elle-même jouant le rôle de base suivant: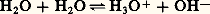 est dite réaction d’autoprotolyse . Elle est caractérisée par la constante:
est dite réaction d’autoprotolyse . Elle est caractérisée par la constante: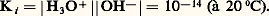 Quantitativité d’une réaction acide-baseLa donnée des valeurs des pKA (tabl. 1) permet de comparer les forces respectives des acides et des bases, et de prévoir le sens prépondérant des réactions. Ainsi de deux couples acide-base de pKA différents, c’est l’acide correspondant à la plus petite valeur de pKA qui sera le plus fort, et c’est donc lui qui réagira sur la base de l’autre couple, et non l’inverse.
Quantitativité d’une réaction acide-baseLa donnée des valeurs des pKA (tabl. 1) permet de comparer les forces respectives des acides et des bases, et de prévoir le sens prépondérant des réactions. Ainsi de deux couples acide-base de pKA différents, c’est l’acide correspondant à la plus petite valeur de pKA qui sera le plus fort, et c’est donc lui qui réagira sur la base de l’autre couple, et non l’inverse.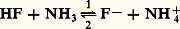 est donc fortement déplacée dans le sens 1. La constante de cet équilibre se déduit facilement des valeurs de KA:
est donc fortement déplacée dans le sens 1. La constante de cet équilibre se déduit facilement des valeurs de KA: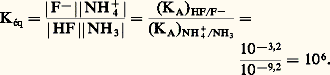 Cette valeur numérique, très grande devant 1, confirme bien les prévisions antérieures d’une réaction très quantitative dans le sens 1. On peut écrire aussi directement:
Cette valeur numérique, très grande devant 1, confirme bien les prévisions antérieures d’une réaction très quantitative dans le sens 1. On peut écrire aussi directement: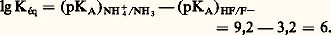 Échelle d’aciditépH d’une solutionLe pH caractérise l’état d’acidité de toute solution contenant des acides et/ou des bases, seuls ou en mélange. Il est défini par la relation:
Échelle d’aciditépH d’une solutionLe pH caractérise l’état d’acidité de toute solution contenant des acides et/ou des bases, seuls ou en mélange. Il est défini par la relation: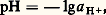 aH+ désignant l’activité des ions hydrogène dans la solution considérée.Échelle de pH dans l’eauL’acidité d’une solution aqueuse résulte de la réaction de transfert de protons d’acides à l’eau et le pH est donné, en confondant en première approximation activité et concentration, par:
aH+ désignant l’activité des ions hydrogène dans la solution considérée.Échelle de pH dans l’eauL’acidité d’une solution aqueuse résulte de la réaction de transfert de protons d’acides à l’eau et le pH est donné, en confondant en première approximation activité et concentration, par: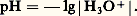 Si l’on convient, pour permettre l’approximation précédente, de limiter toutes les concentrations introduites à 1 mole . l-1, la concentration maximale en H3+ est égale à cette valeur. L’échelle de pH est donc limitée, en pratique, à la valeur 0 en milieu acide.Les acides qui cèdent quantitativement leurs protons à l’eau sont dits acides forts : c’est le cas de HCl4, H3, HCl, H2S4. Dire que la première acidité de l’acide sulfurique est forte traduit le fait que la réaction:
Si l’on convient, pour permettre l’approximation précédente, de limiter toutes les concentrations introduites à 1 mole . l-1, la concentration maximale en H3+ est égale à cette valeur. L’échelle de pH est donc limitée, en pratique, à la valeur 0 en milieu acide.Les acides qui cèdent quantitativement leurs protons à l’eau sont dits acides forts : c’est le cas de HCl4, H3, HCl, H2S4. Dire que la première acidité de l’acide sulfurique est forte traduit le fait que la réaction: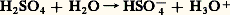 est totale de gauche à droite.Il en résulte que tous les acides forts introduits dans l’eau sont complètement transformés en l’espèce H3+, qui constitue par conséquent l’acide le plus fort qui puisse exister dans l’eau. On notera que ce nivellement des propriétés des acides forts est dû aux propriétés basiques de l’eau.De façon analogue, certaines bases appelées bases fortes (soude, potasse...) ont une affinité pour le proton telle que leur réaction sur l’eau, jouant le rôle d’acide, est quantitative; par exemple, la réaction avec la soude:
est totale de gauche à droite.Il en résulte que tous les acides forts introduits dans l’eau sont complètement transformés en l’espèce H3+, qui constitue par conséquent l’acide le plus fort qui puisse exister dans l’eau. On notera que ce nivellement des propriétés des acides forts est dû aux propriétés basiques de l’eau.De façon analogue, certaines bases appelées bases fortes (soude, potasse...) ont une affinité pour le proton telle que leur réaction sur l’eau, jouant le rôle d’acide, est quantitative; par exemple, la réaction avec la soude: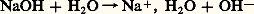 ait apparaître que OH- est la base la plus forte qui puisse être présente en solution aqueuse. Ce sont cette fois les propriétés acides de l’eau qui entraînent un nivellement des propriétés des bases fortes et introduisent une limite supérieure à l’échelle de pH. Pour une base forte mise en solution dans l’eau à la concentration 1 mole . l-1, il y a formation d’ions OH- à la même concentration. De la constante d’autoprotolyse de l’eau, on tire aisément:
ait apparaître que OH- est la base la plus forte qui puisse être présente en solution aqueuse. Ce sont cette fois les propriétés acides de l’eau qui entraînent un nivellement des propriétés des bases fortes et introduisent une limite supérieure à l’échelle de pH. Pour une base forte mise en solution dans l’eau à la concentration 1 mole . l-1, il y a formation d’ions OH- à la même concentration. De la constante d’autoprotolyse de l’eau, on tire aisément: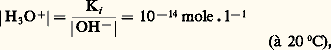 soit:
soit: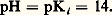 L’échelle de pH est donc limitée à la valeur 14, ou plus généralement pKi , en milieu basique.Dans le cas de l’eau pure, l’équilibre d’autoprotolyse implique:
L’échelle de pH est donc limitée à la valeur 14, ou plus généralement pKi , en milieu basique.Dans le cas de l’eau pure, l’équilibre d’autoprotolyse implique: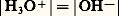 (relation exprimant également l’électroneutralité de la solution). Il vient, par suite:
(relation exprimant également l’électroneutralité de la solution). Il vient, par suite: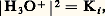 soit:
soit: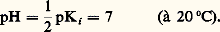 Le milieu est dit acide lorsque |H3+| 礪 |OH- |, soit pH 麗 7; il est dit basique dans le cas contraire, soit pH 礪 7. Il est clair que le pH de la solution d’un acide dans l’eau est au plus égal à 7, et qu’il est d’autant plus faible, à concentration fixée, que l’acide est plus fort et, pour un acide donné, qu’il est plus concentré. Le même raisonnement s’applique, de façon symétrique, aux solutions de bases (pH 礪 7 et d’autant plus élevé que la base est plus forte et plus concentrée).Domaines de prédominancePour tout couple acide-base, de constante d’acidité:
Le milieu est dit acide lorsque |H3+| 礪 |OH- |, soit pH 麗 7; il est dit basique dans le cas contraire, soit pH 礪 7. Il est clair que le pH de la solution d’un acide dans l’eau est au plus égal à 7, et qu’il est d’autant plus faible, à concentration fixée, que l’acide est plus fort et, pour un acide donné, qu’il est plus concentré. Le même raisonnement s’applique, de façon symétrique, aux solutions de bases (pH 礪 7 et d’autant plus élevé que la base est plus forte et plus concentrée).Domaines de prédominancePour tout couple acide-base, de constante d’acidité: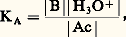 le pH, donné par la relation:
le pH, donné par la relation: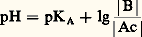 est bien caractéristique de la composition de la solution à l’équilibre, ainsi:
est bien caractéristique de la composition de la solution à l’équilibre, ainsi: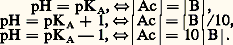 Ces résultats sont représentés sous la forme d’un diagramme de prédominance, montrant que la forme acide est prépondérante lorsque pH 麗 pKA, la forme basique lorsque pH 礪 pKA (fig. 1).Le pH, dont la détermination expérimentale est aisée, permet donc de suivre l’évolution de la composition de la solution, par exemple lors de l’addition d’un réactif titrant.Application à la prévision des réactions acide-base en solution aqueuseSi, sur une échelle de pH (fig. 2), on repère chaque couple acide-base par sa valeur de pKA, c’est-à-dire la valeur de pH d’une solution contenant les deux formes conjuguées à des concentrations égales, l’écart entre deux couples sur l’échelle fournit directement la valeur de lg Kéq = (pKA)2 漣 (pKA)1.En reprenant l’exemple de la réaction:
Ces résultats sont représentés sous la forme d’un diagramme de prédominance, montrant que la forme acide est prépondérante lorsque pH 麗 pKA, la forme basique lorsque pH 礪 pKA (fig. 1).Le pH, dont la détermination expérimentale est aisée, permet donc de suivre l’évolution de la composition de la solution, par exemple lors de l’addition d’un réactif titrant.Application à la prévision des réactions acide-base en solution aqueuseSi, sur une échelle de pH (fig. 2), on repère chaque couple acide-base par sa valeur de pKA, c’est-à-dire la valeur de pH d’une solution contenant les deux formes conjuguées à des concentrations égales, l’écart entre deux couples sur l’échelle fournit directement la valeur de lg Kéq = (pKA)2 漣 (pKA)1.En reprenant l’exemple de la réaction: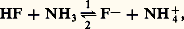 on obtient, par lecture directe sur l’échelle de pH:
on obtient, par lecture directe sur l’échelle de pH: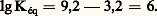 D’où la règle: tout acide réagit sur toute base située au-dessous de lui sur l’échelle, et la réaction est d’autant plus quantitative que l’écart entre les deux couples est plus grand.Sans la limitation par les propriétés respectivement basiques et acides de l’eau, les acides forts figureraient dans la partie supérieure de l’échelle (pH 麗 0), les bases fortes dans la partie inférieure (pH 礪 14).La limitation de l’échelle due au solvant fait que les formes conjuguées des acides et des bases fortes sont respectivement des bases et des acides infiniment faibles, perdant de ce fait toute aptitude à fixer ou céder des protons: ce sont des espèces neutres du point de vue de l’acidité.Ainsi, les ions Cl- (base conjuguée de l’acide fort HCl) et Na+ (acide conjugué de la base forte NaOH) sont des ions neutres: leur présence ne modifie pas le pH de solutions dans lesquelles ils sont introduits.pH des solutions d’électrolyteDe nombreux composés ioniques, comme les sels alcalins et alcalino-terreux, sont entièrement dissociés en solution aqueuse, effet résultant de la solvatation des ions du solide par les molécules d’eau: ce sont des électrolytes forts. Les solutions obtenues peuvent présenter un caractère acide ou basique, selon la nature des ions mis en jeu.Ainsi le fluorure de sodium est totalement dissocié:
D’où la règle: tout acide réagit sur toute base située au-dessous de lui sur l’échelle, et la réaction est d’autant plus quantitative que l’écart entre les deux couples est plus grand.Sans la limitation par les propriétés respectivement basiques et acides de l’eau, les acides forts figureraient dans la partie supérieure de l’échelle (pH 麗 0), les bases fortes dans la partie inférieure (pH 礪 14).La limitation de l’échelle due au solvant fait que les formes conjuguées des acides et des bases fortes sont respectivement des bases et des acides infiniment faibles, perdant de ce fait toute aptitude à fixer ou céder des protons: ce sont des espèces neutres du point de vue de l’acidité.Ainsi, les ions Cl- (base conjuguée de l’acide fort HCl) et Na+ (acide conjugué de la base forte NaOH) sont des ions neutres: leur présence ne modifie pas le pH de solutions dans lesquelles ils sont introduits.pH des solutions d’électrolyteDe nombreux composés ioniques, comme les sels alcalins et alcalino-terreux, sont entièrement dissociés en solution aqueuse, effet résultant de la solvatation des ions du solide par les molécules d’eau: ce sont des électrolytes forts. Les solutions obtenues peuvent présenter un caractère acide ou basique, selon la nature des ions mis en jeu.Ainsi le fluorure de sodium est totalement dissocié: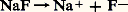 .- est une base, Na+ un ion neutre du point de vue de l’acidité ; la solution se comportera comme celle de la base -.Avec le chlorure d’ammonium:
.- est une base, Na+ un ion neutre du point de vue de l’acidité ; la solution se comportera comme celle de la base -.Avec le chlorure d’ammonium: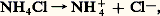 NH4+ est un acide, Cl- un ion neutre du point de vue de l’acidité.Une solution du fluorure d’ammonium:
NH4+ est un acide, Cl- un ion neutre du point de vue de l’acidité.Une solution du fluorure d’ammonium: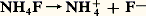 correspond au mélange équimoléculaire de l’acide NH4+ et de la base -; le pH observé est le même que celui qu’on obtient en mélangeant NH3 et HF en proportions stœchiométriques.Une solution d’hydrogénocarbonate de sodium:
correspond au mélange équimoléculaire de l’acide NH4+ et de la base -; le pH observé est le même que celui qu’on obtient en mélangeant NH3 et HF en proportions stœchiométriques.Une solution d’hydrogénocarbonate de sodium: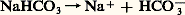 se comporte, du point de vue de l’acido-basicité, comme une solution de l’ampholyte HC3- et son pH est le même que celui d’un mélange équimoléculaire C2, H2O + C32-. Compte tenu de la place des couples HC3-/C32- (pKA1 = 10,3) et C2, H2O/HC3- (pKA2 = 6,4), HCO3- est une forme stable; la réaction:
se comporte, du point de vue de l’acido-basicité, comme une solution de l’ampholyte HC3- et son pH est le même que celui d’un mélange équimoléculaire C2, H2O + C32-. Compte tenu de la place des couples HC3-/C32- (pKA1 = 10,3) et C2, H2O/HC3- (pKA2 = 6,4), HCO3- est une forme stable; la réaction: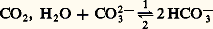 est déplacée dans le sens 1 de l’amphotérisation (la réaction inverse, pratiquement inexistante ici, est dite de dismutation).Échelles de pH en milieu non aqueuxConsidérons un solvant amphiprotique HS. Le nivellement des propriétés des acides forts dans ce solvant résulte de son caractère basique plus ou moins prononcé, c’est-à-dire de son affinité pour la particule H+ (couple H2S+/HS). On conçoit que, dans un solvant moins basique que l’eau, on puisse observer une différenciation dans la force de ces acides, qui se comporteront alors comme des acides faibles de constantes KA différentes:
est déplacée dans le sens 1 de l’amphotérisation (la réaction inverse, pratiquement inexistante ici, est dite de dismutation).Échelles de pH en milieu non aqueuxConsidérons un solvant amphiprotique HS. Le nivellement des propriétés des acides forts dans ce solvant résulte de son caractère basique plus ou moins prononcé, c’est-à-dire de son affinité pour la particule H+ (couple H2S+/HS). On conçoit que, dans un solvant moins basique que l’eau, on puisse observer une différenciation dans la force de ces acides, qui se comporteront alors comme des acides faibles de constantes KA différentes: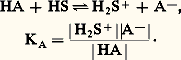 Dans un tel solvant, où l’acide le plus fort pouvant être effectivement présent en solution est H2S+, la limite inférieure de l’échelle de pH, toujours définie par |H2S+| = 1 mole . l-1, soit pH = 0, correspond à un milieu plus acide que ceux qui sont accessibles dans l’eau; on peut dire également que cette origine de l’échelle dans HS équivaut à un pH négatif sur l’échelle dans l’eau. Des solvants très peu basiques, comme l’acide fluorosulfurique, permettent d’atteindre des milieux extrêmement acides. On peut y observer des réactions inconnues dans l’eau.Par un raisonnement analogue, on prévoit que des milieux très basiques pourront être obtenus avec des solvants très peu acides (HS très faiblement donneur de protons dans le couple HS/S-). Ainsi, l’ammoniac liquide permet d’atteindre l’équivalent de pH = 40 ramené à l’échelle dans l’eau.Détermination du pH d’une solution. Titrages acido-basiquesDétermination expérimentale du pHMesure du pH (pH-métrie)Le pH est couramment déterminé par une mesure de force électromotrice. On constitue une pile comportant une électrode de potentiel constant (électrode de référence) et une électrode de verre, dont le potentiel dépend du pH de la solution (électrode indicatrice). Cette seconde électrode comprend une solution de remplissage interne de pH0 connu, dans laquelle est immergée une seconde électrode de référence. L’échange d’ions intervenant sur les deux faces de la membrane de verre entre les ions hydrogène et les ions sodium entrant dans la composition du verre conduit à l’apparition d’un potentiel de membrane, fonction affine de la différence (pH0 漣 pH), de telle sorte que la différence de potentiel mesurée entre les deux électrodes est de la forme:
Dans un tel solvant, où l’acide le plus fort pouvant être effectivement présent en solution est H2S+, la limite inférieure de l’échelle de pH, toujours définie par |H2S+| = 1 mole . l-1, soit pH = 0, correspond à un milieu plus acide que ceux qui sont accessibles dans l’eau; on peut dire également que cette origine de l’échelle dans HS équivaut à un pH négatif sur l’échelle dans l’eau. Des solvants très peu basiques, comme l’acide fluorosulfurique, permettent d’atteindre des milieux extrêmement acides. On peut y observer des réactions inconnues dans l’eau.Par un raisonnement analogue, on prévoit que des milieux très basiques pourront être obtenus avec des solvants très peu acides (HS très faiblement donneur de protons dans le couple HS/S-). Ainsi, l’ammoniac liquide permet d’atteindre l’équivalent de pH = 40 ramené à l’échelle dans l’eau.Détermination du pH d’une solution. Titrages acido-basiquesDétermination expérimentale du pHMesure du pH (pH-métrie)Le pH est couramment déterminé par une mesure de force électromotrice. On constitue une pile comportant une électrode de potentiel constant (électrode de référence) et une électrode de verre, dont le potentiel dépend du pH de la solution (électrode indicatrice). Cette seconde électrode comprend une solution de remplissage interne de pH0 connu, dans laquelle est immergée une seconde électrode de référence. L’échange d’ions intervenant sur les deux faces de la membrane de verre entre les ions hydrogène et les ions sodium entrant dans la composition du verre conduit à l’apparition d’un potentiel de membrane, fonction affine de la différence (pH0 漣 pH), de telle sorte que la différence de potentiel mesurée entre les deux électrodes est de la forme: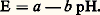 L’étalonnage du pH-mètre, qui est un millivoltmètre électronique permettant une lecture directe en pH, correspond de fait à la détermination expérimentale des constantes a et b au moyen de solutions de pH connu (il existe des solutions étalons de pH préconisées par le National Bureau of Standards, utilisables en milieu aqueux).Repérage du pH. Indicateurs colorésLe pH des solutions peut également être déterminé de façon semi-quantitative au moyen d’indicateurs colorés de pH: ce sont des couples acide-base dont chacune des deux formes conjuguées présente une coloration différente et intense (colorants organiques). Introduits en très faible concentration, ils adoptent le pH de la solution, ce qui fixe le rapport |IB|/|IAc| des concentrations des deux formes. On considère, en règle générale, que l’on n’observe à l’œil qu’une seule coloration lorsque l’une des deux espèces est dix fois plus concentrée que l’autre. Il en résulte que l’intervalle de virage de chaque indicateur s’étend sur deux unités de pH environ autour du pKA de l’indicateur (fig. 2). Ainsi, le rouge de méthyle vire du rouge au jaune sur l’intervalle 4,2-6,2.Utilisés directement en solution, les indicateurs colorés permettent de détecter des variations brutales de pH; bien choisis, ils peuvent servir au repérage des points d’équivalence, au cours de titrages volumétriques. Imprégnés en mélange sur des bandelettes de papier, que l’on plonge dans les solutions, ils permettent un repérage du pH par comparaison avec un échelle de teintes (papiers indicateurs de pH).Calcul du pH d’une solutionL’état d’une solution est entièrement défini lorsqu’on a exprimé les différentes constantes et les bilans de conservation des espèces. Le pH de toute solution peut donc être calculé a priori.Prenons l’exemple d’un mélange du monoacide HA et de sa base conjuguée A-, introduits respectivement à la concentration CHA et CA- dans un solvant amphiprotique HS. On montre facilement que le pH de la solution (face=F0019 漣 lg|H2S+|) se déduit de l’équation:
L’étalonnage du pH-mètre, qui est un millivoltmètre électronique permettant une lecture directe en pH, correspond de fait à la détermination expérimentale des constantes a et b au moyen de solutions de pH connu (il existe des solutions étalons de pH préconisées par le National Bureau of Standards, utilisables en milieu aqueux).Repérage du pH. Indicateurs colorésLe pH des solutions peut également être déterminé de façon semi-quantitative au moyen d’indicateurs colorés de pH: ce sont des couples acide-base dont chacune des deux formes conjuguées présente une coloration différente et intense (colorants organiques). Introduits en très faible concentration, ils adoptent le pH de la solution, ce qui fixe le rapport |IB|/|IAc| des concentrations des deux formes. On considère, en règle générale, que l’on n’observe à l’œil qu’une seule coloration lorsque l’une des deux espèces est dix fois plus concentrée que l’autre. Il en résulte que l’intervalle de virage de chaque indicateur s’étend sur deux unités de pH environ autour du pKA de l’indicateur (fig. 2). Ainsi, le rouge de méthyle vire du rouge au jaune sur l’intervalle 4,2-6,2.Utilisés directement en solution, les indicateurs colorés permettent de détecter des variations brutales de pH; bien choisis, ils peuvent servir au repérage des points d’équivalence, au cours de titrages volumétriques. Imprégnés en mélange sur des bandelettes de papier, que l’on plonge dans les solutions, ils permettent un repérage du pH par comparaison avec un échelle de teintes (papiers indicateurs de pH).Calcul du pH d’une solutionL’état d’une solution est entièrement défini lorsqu’on a exprimé les différentes constantes et les bilans de conservation des espèces. Le pH de toute solution peut donc être calculé a priori.Prenons l’exemple d’un mélange du monoacide HA et de sa base conjuguée A-, introduits respectivement à la concentration CHA et CA- dans un solvant amphiprotique HS. On montre facilement que le pH de la solution (face=F0019 漣 lg|H2S+|) se déduit de l’équation: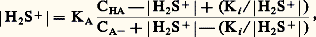 Ki et KA désignant respectivement le produit ionique du solvant HS et la constante d’acidité du couple HA/A- dans HS.Cette équation est du troisième degré en |H2S+| et l’on conçoit que le degré augmente rapidement avec le nombre d’équilibres mis en jeu, par exemple dans le cas de polyacides. L’informatique permet le calcul du pH pour toute solution de composition connue, et par suite la simulation des courbes de titrages (on appelle ainsi la courbe représentant les variations du pH d’une solution en fonction du volume de réactif titrant introduit). Considérons, à titre d’exemple, la réaction de titrage d’un acide faible HA par une base forte, en solution aqueuse:
Ki et KA désignant respectivement le produit ionique du solvant HS et la constante d’acidité du couple HA/A- dans HS.Cette équation est du troisième degré en |H2S+| et l’on conçoit que le degré augmente rapidement avec le nombre d’équilibres mis en jeu, par exemple dans le cas de polyacides. L’informatique permet le calcul du pH pour toute solution de composition connue, et par suite la simulation des courbes de titrages (on appelle ainsi la courbe représentant les variations du pH d’une solution en fonction du volume de réactif titrant introduit). Considérons, à titre d’exemple, la réaction de titrage d’un acide faible HA par une base forte, en solution aqueuse: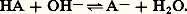 La figure 3 montre les courbes simulées, à concentration donnée et pKA variable.Il existe des cas où des approximations sont possibles, conduisant aux «formules simplifiées de pH», dont les principales sont rassemblées dans le tableau 2. Si elles permettent à l’utilisateur une évaluation rapide du pH, leur application implique que la validité des approximations mises en jeu ait été vérifiée. Ainsi, par dilution, un acide faible, de pKA 諒 5, peut se comporter comme un acide fort, conséquence de la loi du déplacement des équilibres par un excès de réactif.Solutions-tamponsConsidérons, sur la figure 3, les variations très lentes du pH de la solution d’acides HA de pKA compris entre 3 et 11, au voisinage de la demi-équivalence (l’équivalence correspond à l’ajout du réactif titrant en proportions stœchiométriques). Une solution contenant l’acide et la base du même couple en concentrations voisines et pas trop faibles constitue une solution-tampon de pH: on peut y ajouter un acide ou une base en quantité assez grande sans que le pH varie notablement. Ainsi, le pH sanguin est tamponné à une valeur proche de 7,4 par plusieurs systèmes, dont H2P4-/HP42-.Dans la suite, pour alléger l’écriture, le proton solvaté sera noté H+.Titrages acido-basiquesLe problème se pose souvent de l’analyse quantitative d’une solution, c’est-à-dire de la détermination de la concentration des espèces. Si celles-ci présentent des propriétés acides ou basiques, on peut opérer par titrage ; on effectue un choix des conditions opératoires (nature du solvant et du réactif titrant) conduisant à une réaction fortement quantitative, condition nécessaire à l’observation d’un saut net de pH autour du point d’équivalence. Cette recherche justifie l’emploi d’acides et de bases fortes dans le solvant choisi comme réactifs titrants et la mise en œuvre de solvants autres que l’eau pour le titrage d’acides et de bases trop faibles dans ce milieu pour y être titrables avec précision.2. Réactions d’oxydoréduction (transfert d’électrons)DéfinitionsLes réactions d’oxydoréduction sont des réactions d’échange d’électrons faisant intervenir des accepteurs et des donneurs de cette particule: les oxydants peuvent fixer des électrons, les réducteurs peuvent en céder. La relation qui définit un oxydant, un réducteur, ou plus globalement un couple oxydoréducteur (en abrégé couple redox) est:
La figure 3 montre les courbes simulées, à concentration donnée et pKA variable.Il existe des cas où des approximations sont possibles, conduisant aux «formules simplifiées de pH», dont les principales sont rassemblées dans le tableau 2. Si elles permettent à l’utilisateur une évaluation rapide du pH, leur application implique que la validité des approximations mises en jeu ait été vérifiée. Ainsi, par dilution, un acide faible, de pKA 諒 5, peut se comporter comme un acide fort, conséquence de la loi du déplacement des équilibres par un excès de réactif.Solutions-tamponsConsidérons, sur la figure 3, les variations très lentes du pH de la solution d’acides HA de pKA compris entre 3 et 11, au voisinage de la demi-équivalence (l’équivalence correspond à l’ajout du réactif titrant en proportions stœchiométriques). Une solution contenant l’acide et la base du même couple en concentrations voisines et pas trop faibles constitue une solution-tampon de pH: on peut y ajouter un acide ou une base en quantité assez grande sans que le pH varie notablement. Ainsi, le pH sanguin est tamponné à une valeur proche de 7,4 par plusieurs systèmes, dont H2P4-/HP42-.Dans la suite, pour alléger l’écriture, le proton solvaté sera noté H+.Titrages acido-basiquesLe problème se pose souvent de l’analyse quantitative d’une solution, c’est-à-dire de la détermination de la concentration des espèces. Si celles-ci présentent des propriétés acides ou basiques, on peut opérer par titrage ; on effectue un choix des conditions opératoires (nature du solvant et du réactif titrant) conduisant à une réaction fortement quantitative, condition nécessaire à l’observation d’un saut net de pH autour du point d’équivalence. Cette recherche justifie l’emploi d’acides et de bases fortes dans le solvant choisi comme réactifs titrants et la mise en œuvre de solvants autres que l’eau pour le titrage d’acides et de bases trop faibles dans ce milieu pour y être titrables avec précision.2. Réactions d’oxydoréduction (transfert d’électrons)DéfinitionsLes réactions d’oxydoréduction sont des réactions d’échange d’électrons faisant intervenir des accepteurs et des donneurs de cette particule: les oxydants peuvent fixer des électrons, les réducteurs peuvent en céder. La relation qui définit un oxydant, un réducteur, ou plus globalement un couple oxydoréducteur (en abrégé couple redox) est: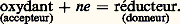 Par exemple,
Par exemple,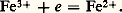 Le passage des métaux en solution constitue un phénomène d’oxydoréduction:
Le passage des métaux en solution constitue un phénomène d’oxydoréduction: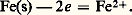 De même, le dépôt d’un métal à partir d’une solution de l’un de ses ions correspond à une réduction:
De même, le dépôt d’un métal à partir d’une solution de l’un de ses ions correspond à une réduction: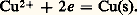 Les ions H+ et l’oxygène dissous, qui interviennent dans les phénomènes de corrosion, sont des oxydants:
Les ions H+ et l’oxygène dissous, qui interviennent dans les phénomènes de corrosion, sont des oxydants: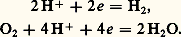 Un composé peut être à la fois oxydant dans un couple et réducteur dans un autre. On l’appelle alors ampholyte ; par exemple, le chlore Cl2 est oxydant dans le couple Cl2/Cl- et réducteur dans le couple HClO/Cl2:
Un composé peut être à la fois oxydant dans un couple et réducteur dans un autre. On l’appelle alors ampholyte ; par exemple, le chlore Cl2 est oxydant dans le couple Cl2/Cl- et réducteur dans le couple HClO/Cl2: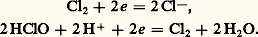 Réactions d’oxydoréductionL’échange d’électrons peut s’effectuer de deux manières, soit en solution, soit à la surface d’électrodes, conducteurs électroniques [cf. ÉLECTROCHIMIE ET ÉLECTROLYSE].Les électrons ne pouvant exister à l’état libre en solution, en particulier aqueuse, pour qu’un oxydant Ox1 d’un couple redox puisse fixer des électrons il faut qu’il soit en présence d’un réducteur Red2 appartenant à un autre couple et capable de les lui céder. Entre les deux couples:
Réactions d’oxydoréductionL’échange d’électrons peut s’effectuer de deux manières, soit en solution, soit à la surface d’électrodes, conducteurs électroniques [cf. ÉLECTROCHIMIE ET ÉLECTROLYSE].Les électrons ne pouvant exister à l’état libre en solution, en particulier aqueuse, pour qu’un oxydant Ox1 d’un couple redox puisse fixer des électrons il faut qu’il soit en présence d’un réducteur Red2 appartenant à un autre couple et capable de les lui céder. Entre les deux couples: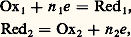 on a la réaction d’oxydoréduction :
on a la réaction d’oxydoréduction :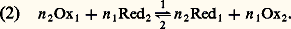 Par exemple, les ions ferreux en présence de dichromate Cr272-, dans lequel le chrome est au degré d’oxydation (D.O.) VI+, cèdent leurs électrons (la considération des degrés d’oxydation permet de déterminer le nombre d’électrons échangés dans un système redox complexe). Les ions Fe2+ sont oxydés en Fe3+, le dichromate est réduit en Cr3+:
Par exemple, les ions ferreux en présence de dichromate Cr272-, dans lequel le chrome est au degré d’oxydation (D.O.) VI+, cèdent leurs électrons (la considération des degrés d’oxydation permet de déterminer le nombre d’électrons échangés dans un système redox complexe). Les ions Fe2+ sont oxydés en Fe3+, le dichromate est réduit en Cr3+: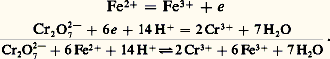 C’est ainsi qu’on «équilibre» une réaction d’oxydoréduction: les électrons ne doivent plus figurer; les deux membres de la réaction doivent comporter le même nombre d’atomes de chaque type et la même charge globale.L’éthanal (aldéhyde acétique) est oxydable en éthanoïque (acide acétique) par le permanganate:
C’est ainsi qu’on «équilibre» une réaction d’oxydoréduction: les électrons ne doivent plus figurer; les deux membres de la réaction doivent comporter le même nombre d’atomes de chaque type et la même charge globale.L’éthanal (aldéhyde acétique) est oxydable en éthanoïque (acide acétique) par le permanganate: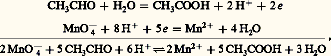 Prévision des réactionsSoit l’équilibre d’oxydoréduction (2): il est important, en pratique, de savoir s’il est déplacé dans le sens 1 (oxydation de Red2 par Ox1; Ox1 étant alors dit plus fort que Ox2 ou Red2 plus fort que Red1) ou dans le sens 2 (oxydation de Red1 par Ox2).Cette prévision peut se faire à l’aide du potentiel d’oxydoréduction de la solution.Potentiel d’oxydoréduction d’une solutionSystème simple. Potentiel normalConsidérons une solution contenant l’oxydant et le réducteur du système:
Prévision des réactionsSoit l’équilibre d’oxydoréduction (2): il est important, en pratique, de savoir s’il est déplacé dans le sens 1 (oxydation de Red2 par Ox1; Ox1 étant alors dit plus fort que Ox2 ou Red2 plus fort que Red1) ou dans le sens 2 (oxydation de Red1 par Ox2).Cette prévision peut se faire à l’aide du potentiel d’oxydoréduction de la solution.Potentiel d’oxydoréduction d’une solutionSystème simple. Potentiel normalConsidérons une solution contenant l’oxydant et le réducteur du système: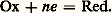 Le potentiel d’équilibre de cette solution est donné par la relation de Nernst:
Le potentiel d’équilibre de cette solution est donné par la relation de Nernst: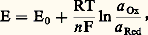 dans laquelle R, T et F désignent respectivement la constante des gaz parfaits (8,32 J . mole-1 . K-1), la température absolue (K) et le faraday (96 485 coulombs); n est le nombre d’électrons mis en jeu, E0 le potentiel normal du système oxydoréducteur considéré, grandeur thermodynamique caractéristique de celui-ci (comme toute grandeur de cette nature, E0 est défini par rapport à une origine arbitraire); par convention, le potentiel normal E0 = 0 V est attribué au système H+/H2 (tabl. 3).a Ox et a Red sont les activités de l’oxydant et du réducteur. Comme il est d’usage en chimie des solutions, nous utiliserons les conventions et approximations suivantes:– l’activité des solutés, présents en solution diluée (face=F0019 麗 1 mol . l-1), est assimilée à leur concentration (notée | | ); en fait, ai = 塚i Ci , avec 塚i coefficient d’activité tendant vers 1 quand C0;– l’activité du solvant, de même que celle des solides (métaux, précipités), est par convention égale à l’unité;– l’activité des gaz est assimilée à leur pression partielle au-dessus de la solution.À température ambiante, ou voisine, on a de plus:
dans laquelle R, T et F désignent respectivement la constante des gaz parfaits (8,32 J . mole-1 . K-1), la température absolue (K) et le faraday (96 485 coulombs); n est le nombre d’électrons mis en jeu, E0 le potentiel normal du système oxydoréducteur considéré, grandeur thermodynamique caractéristique de celui-ci (comme toute grandeur de cette nature, E0 est défini par rapport à une origine arbitraire); par convention, le potentiel normal E0 = 0 V est attribué au système H+/H2 (tabl. 3).a Ox et a Red sont les activités de l’oxydant et du réducteur. Comme il est d’usage en chimie des solutions, nous utiliserons les conventions et approximations suivantes:– l’activité des solutés, présents en solution diluée (face=F0019 麗 1 mol . l-1), est assimilée à leur concentration (notée | | ); en fait, ai = 塚i Ci , avec 塚i coefficient d’activité tendant vers 1 quand C0;– l’activité du solvant, de même que celle des solides (métaux, précipités), est par convention égale à l’unité;– l’activité des gaz est assimilée à leur pression partielle au-dessus de la solution.À température ambiante, ou voisine, on a de plus: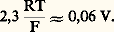 La relation de Nernst s’écrit alors:
La relation de Nernst s’écrit alors: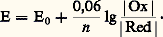 Par exemple, on obtient, pour les systèmes suivants:
Par exemple, on obtient, pour les systèmes suivants: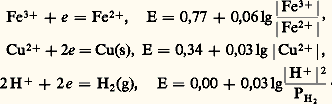 Influence du milieu: potentiel normal apparentPour de nombreux couples redox, l’électron n’est pas la seule particule mise en jeu et le milieu influe sur les propriétés oxydoréductrices. Il est commode d’introduire alors la notion de potentiel normal apparent .Considérons, par exemple, le système redox Mn4-/Mn2+:
Influence du milieu: potentiel normal apparentPour de nombreux couples redox, l’électron n’est pas la seule particule mise en jeu et le milieu influe sur les propriétés oxydoréductrices. Il est commode d’introduire alors la notion de potentiel normal apparent .Considérons, par exemple, le système redox Mn4-/Mn2+: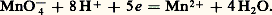 La loi de Nernst s’écrit avec les approximations vues précédemment:
La loi de Nernst s’écrit avec les approximations vues précédemment: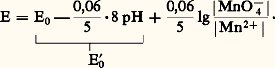 On remarque que, si on remplace E0 par E 0 (potentiel normal apparent), on retrouve l’écriture de la loi de Nernst dans le cas d’un système simple. E 0 dépend linéairement du pH d’une manière qui découle simplement de l’écriture du couple (nombre de H+ et nombre d’électrons). Pour les systèmes de ce type, les tables de constantes fournissent les valeurs du potentiel normal apparent à un pH donné, généralement pH 0. Dans le cas du couple Mn4-/Mn2+, on relève la valeur 1,50 V. On a alors:
On remarque que, si on remplace E0 par E 0 (potentiel normal apparent), on retrouve l’écriture de la loi de Nernst dans le cas d’un système simple. E 0 dépend linéairement du pH d’une manière qui découle simplement de l’écriture du couple (nombre de H+ et nombre d’électrons). Pour les systèmes de ce type, les tables de constantes fournissent les valeurs du potentiel normal apparent à un pH donné, généralement pH 0. Dans le cas du couple Mn4-/Mn2+, on relève la valeur 1,50 V. On a alors: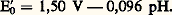 Mesure du potentiel redox. Électrode de référenceLorsque, dans une solution contenant un couple redox, on plonge un fil inattaquable (par exemple un fil de platine), ce dernier, par suite d’un échange incessant d’électrons avec les corps dissous, prend le potentiel d’équilibre de la solution. Mais, comme seules les différences de potentiel sont mesurables, on doit disposer d’une seconde électrode de potentiel fixe (électrode de référence).Électrode normale à hydrogène (E.N.H.). Elle est constituée d’une plaque de platine, platinée par électrolyse, immergée dans une solution d’un acide fort à pH 0 et saturée d’hydrogène sous la pression de 1 atmosphère. Elle prend le potentiel donné par la relation de Nernst pour le couple:
Mesure du potentiel redox. Électrode de référenceLorsque, dans une solution contenant un couple redox, on plonge un fil inattaquable (par exemple un fil de platine), ce dernier, par suite d’un échange incessant d’électrons avec les corps dissous, prend le potentiel d’équilibre de la solution. Mais, comme seules les différences de potentiel sont mesurables, on doit disposer d’une seconde électrode de potentiel fixe (électrode de référence).Électrode normale à hydrogène (E.N.H.). Elle est constituée d’une plaque de platine, platinée par électrolyse, immergée dans une solution d’un acide fort à pH 0 et saturée d’hydrogène sous la pression de 1 atmosphère. Elle prend le potentiel donné par la relation de Nernst pour le couple: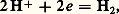 soit:
soit: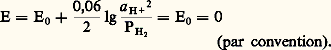 Cette électrode est peu maniable et n’est utilisée que par des spécialistes.Électrode au calomel, KCl saturé (E.C.S.). Son fonctionnement est fondé sur les propriétés du couple Hg(I)/Hg en milieu chlorure:
Cette électrode est peu maniable et n’est utilisée que par des spécialistes.Électrode au calomel, KCl saturé (E.C.S.). Son fonctionnement est fondé sur les propriétés du couple Hg(I)/Hg en milieu chlorure: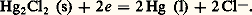 Un conducteur inattaquable plongeant dans du mercure au contact de calomel Hg2Cl2 (très peu soluble) adopte le potentiel:
Un conducteur inattaquable plongeant dans du mercure au contact de calomel Hg2Cl2 (très peu soluble) adopte le potentiel: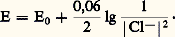 Son potentiel est donc constant si la concentration en chlorure est maintenue elle-même constante. Cela peut être obtenu en saturant la solution en un chlorure très soluble, KCl par exemple. Dans ce cas, le potentiel obtenu est 0,241 VE.N.H. à 20 0C.Autres électrodes de référence . Parmi les électrodes de référence usuelles, citons encore l’électrode Ag/AgCl (E = 0,197 VE.N.H.), très facilement miniaturisable, et l’électrode au sulfate mercureux, K2S4 saturé (E = 0,64 V/E.N.H.).Prévision des réactions d’oxydoréduction. Échelle redoxCas des systèmes simplesConsidérons les deux couples oxydoréducteurs simples:
Son potentiel est donc constant si la concentration en chlorure est maintenue elle-même constante. Cela peut être obtenu en saturant la solution en un chlorure très soluble, KCl par exemple. Dans ce cas, le potentiel obtenu est 0,241 VE.N.H. à 20 0C.Autres électrodes de référence . Parmi les électrodes de référence usuelles, citons encore l’électrode Ag/AgCl (E = 0,197 VE.N.H.), très facilement miniaturisable, et l’électrode au sulfate mercureux, K2S4 saturé (E = 0,64 V/E.N.H.).Prévision des réactions d’oxydoréduction. Échelle redoxCas des systèmes simplesConsidérons les deux couples oxydoréducteurs simples: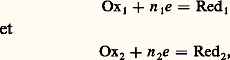 de potentiels normaux respectifs E01 et E02, et l’équilibre de transfert d’électrons auquel ils peuvent donner lieu:
de potentiels normaux respectifs E01 et E02, et l’équilibre de transfert d’électrons auquel ils peuvent donner lieu: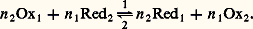 Cet équilibre est caractérisé par la constante:
Cet équilibre est caractérisé par la constante: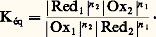 Or le potentiel d’oxydoréduction d’une solution est unique et l’on a, à l’équilibre:
Or le potentiel d’oxydoréduction d’une solution est unique et l’on a, à l’équilibre: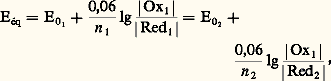 d’où l’on tire simplement:
d’où l’on tire simplement: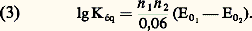 Le sens prépondérant de la réaction est le sens 1 ou 2 selon que la constante Kéq est grande ou, au contraire, petite devant 1, d’où la règle: pour que l’oxydant d’un système puisse réagir avec le réducteur d’un autre système, il faut que le potentiel normal du premier couple soit supérieur à celui du second, et la réaction a d’autant plus de chances d’être quantitative que l’écart est plus important.Il est commode pour appliquer cette règle de représenter les différents systèmes sur une échelle de potentiel, chaque couple y étant repéré par la valeur de son potentiel normal, c’est-à-dire le potentiel d’une solution contenant l’oxydant et le réducteur à la même concentration (fig. 4). Ainsi le zinc est attaqué par les acides forts et non le cuivre (dans le cas où H+ est l’oxydant mis en jeu), et une lame de zinc plongée dans une solution de Cu2+ se recouvre de cuivre métallique.Cas des systèmes complexesDans ce cas, la prévision des réactions se fait avec les potentiels normaux apparents. On les introduit à la place des potentiels normaux dans la relation (3). Le sens et la quantitativité de la réaction dépendent du pH, puisque les potentiels normaux apparents en dépendent eux-mêmes. En pratique, il suffit de superposer sur un même diagramme les variations, en fonction du pH, des potentiels normaux apparents des deux couples et d’appliquer la règle édictée précédemment.Par exemple, les propriétés redox du couple I2/I- sont indépendantes du pH (fig. 5):
Le sens prépondérant de la réaction est le sens 1 ou 2 selon que la constante Kéq est grande ou, au contraire, petite devant 1, d’où la règle: pour que l’oxydant d’un système puisse réagir avec le réducteur d’un autre système, il faut que le potentiel normal du premier couple soit supérieur à celui du second, et la réaction a d’autant plus de chances d’être quantitative que l’écart est plus important.Il est commode pour appliquer cette règle de représenter les différents systèmes sur une échelle de potentiel, chaque couple y étant repéré par la valeur de son potentiel normal, c’est-à-dire le potentiel d’une solution contenant l’oxydant et le réducteur à la même concentration (fig. 4). Ainsi le zinc est attaqué par les acides forts et non le cuivre (dans le cas où H+ est l’oxydant mis en jeu), et une lame de zinc plongée dans une solution de Cu2+ se recouvre de cuivre métallique.Cas des systèmes complexesDans ce cas, la prévision des réactions se fait avec les potentiels normaux apparents. On les introduit à la place des potentiels normaux dans la relation (3). Le sens et la quantitativité de la réaction dépendent du pH, puisque les potentiels normaux apparents en dépendent eux-mêmes. En pratique, il suffit de superposer sur un même diagramme les variations, en fonction du pH, des potentiels normaux apparents des deux couples et d’appliquer la règle édictée précédemment.Par exemple, les propriétés redox du couple I2/I- sont indépendantes du pH (fig. 5):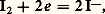 avec:
avec: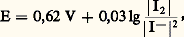 tandis que celles du couple I3-/I2 en dépendent:
tandis que celles du couple I3-/I2 en dépendent: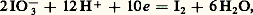 avec:
avec: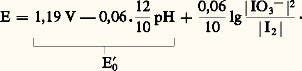 La figure 5 montre que, en dessous de pH 7,9, I3- peut oxyder I- selon la réaction, appelée amphotérisation :
La figure 5 montre que, en dessous de pH 7,9, I3- peut oxyder I- selon la réaction, appelée amphotérisation :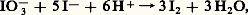 et ce d’autant plus quantitativement que le pH est plus faible. Au-dessus de pH 7,9, I2 oxydant oxyde I2 réducteur (I2 est un ampholyte ), selon la réaction inverse de la précédente:
et ce d’autant plus quantitativement que le pH est plus faible. Au-dessus de pH 7,9, I2 oxydant oxyde I2 réducteur (I2 est un ampholyte ), selon la réaction inverse de la précédente: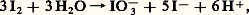 appelée dismutation et qui traduit une instabilité thermodynamique de l’iode dans les conditions indiquées. Dans ce domaine de pH, on réduit directement I3- en I- selon:
appelée dismutation et qui traduit une instabilité thermodynamique de l’iode dans les conditions indiquées. Dans ce domaine de pH, on réduit directement I3- en I- selon: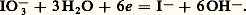 Vitesse des réactions d’oxydoréductionNous venons de voir comment les potentiels normaux, ou normaux apparents, permettaient de prévoir la constante d’équilibre d’une réaction d’oxydoréduction. Mentionnons toutefois la faiblesse des prévisions purement thermodynamiques: elles n’indiquent que la possibilité des réactions, sans présumer de leur vitesse. Elles ne permettent pas, par exemple, d’expliquer pourquoi l’aluminium très pur n’est attaqué que très lentement par l’acide sulfurique dilué alors que, d’après le potentiel normal du couple Al3+/Al(s) (E0 = 漣 1,66 V), on s’attend à une réaction très quantitative.Il est difficile de prévoir la vitesse des réactions. En pratique, elle dépend de la température, des concentrations, de l’écart des potentiels redox, de la nature des couples mis en présence. Certains oxydants ou réducteurs agissent toujours lentement. En règle générale, lorsque l’échange d’électrons n’engendre pas de changement de structure considérable, les réactions sont rapides. C’est le cas, par exemple, des systèmes fer ferriqueer ferreux ou ferricyanureerrocyanure:
Vitesse des réactions d’oxydoréductionNous venons de voir comment les potentiels normaux, ou normaux apparents, permettaient de prévoir la constante d’équilibre d’une réaction d’oxydoréduction. Mentionnons toutefois la faiblesse des prévisions purement thermodynamiques: elles n’indiquent que la possibilité des réactions, sans présumer de leur vitesse. Elles ne permettent pas, par exemple, d’expliquer pourquoi l’aluminium très pur n’est attaqué que très lentement par l’acide sulfurique dilué alors que, d’après le potentiel normal du couple Al3+/Al(s) (E0 = 漣 1,66 V), on s’attend à une réaction très quantitative.Il est difficile de prévoir la vitesse des réactions. En pratique, elle dépend de la température, des concentrations, de l’écart des potentiels redox, de la nature des couples mis en présence. Certains oxydants ou réducteurs agissent toujours lentement. En règle générale, lorsque l’échange d’électrons n’engendre pas de changement de structure considérable, les réactions sont rapides. C’est le cas, par exemple, des systèmes fer ferriqueer ferreux ou ferricyanureerrocyanure: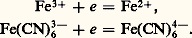 Ce n’est pas le cas du système persulfate/sulfate:
Ce n’est pas le cas du système persulfate/sulfate: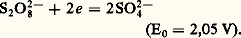 Le persulfate qui, d’après ce potentiel normal, est un oxydant très énergique a une action, dans la plupart des cas, si lente qu’on peut la tenir pour négligeable en l’absence de catalyseur.Rôle du solvantLe solvant et ses ions peuvent échanger des électrons. Ainsi, l’eau peut jouer le rôle d’oxydant:
Le persulfate qui, d’après ce potentiel normal, est un oxydant très énergique a une action, dans la plupart des cas, si lente qu’on peut la tenir pour négligeable en l’absence de catalyseur.Rôle du solvantLe solvant et ses ions peuvent échanger des électrons. Ainsi, l’eau peut jouer le rôle d’oxydant: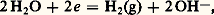 avec E = 漣 0,06 pH si PH2 = 1 atm, ou de réducteur:
avec E = 漣 0,06 pH si PH2 = 1 atm, ou de réducteur: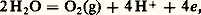 avec E = 1,23 V 漣 0,06 pH si PO2 = 1 atm. Les variations, en fonction du pH, des potentiels normaux apparents de ces deux couples redox de l’eau sont représentés sur la figure 5 (courbes a et b).En principe, tout oxydant et tout réducteur appartenant à des couples dont le potentiel normal apparent est situé respectivement au-dessus de (a) ou au-dessous de (b) devraient oxyder ou réduire l’eau et, par conséquent, ne pas exister.De façon générale, à température ordinaire, en l’absence de catalyseur, les réactions mettant en jeu l’eau sont très lentes. À pH 0, par exemple, Mn4- (E 0 = 1,68 V dans le couple Mn4-/Mn2), Ce4+ (E 0 = 1,70 V dans le couple Ce4+/Ce3+ en milieu perchlorate) ou Cr2+ (E 0 = 漣 0,41 V dans le couple Cr3+/Cr2+) existent en faux équilibre.Dans le cas d’oxydants ou de réducteurs extrêmement énergiques, les réactions sur l’eau sont rapides. C’est le cas, par exemple, du fluor (E0 = 2,87 V) ou du sodium (E0 = 漣 2,71 V) qui réagissent quantitativement sur l’eau selon:
avec E = 1,23 V 漣 0,06 pH si PO2 = 1 atm. Les variations, en fonction du pH, des potentiels normaux apparents de ces deux couples redox de l’eau sont représentés sur la figure 5 (courbes a et b).En principe, tout oxydant et tout réducteur appartenant à des couples dont le potentiel normal apparent est situé respectivement au-dessus de (a) ou au-dessous de (b) devraient oxyder ou réduire l’eau et, par conséquent, ne pas exister.De façon générale, à température ordinaire, en l’absence de catalyseur, les réactions mettant en jeu l’eau sont très lentes. À pH 0, par exemple, Mn4- (E 0 = 1,68 V dans le couple Mn4-/Mn2), Ce4+ (E 0 = 1,70 V dans le couple Ce4+/Ce3+ en milieu perchlorate) ou Cr2+ (E 0 = 漣 0,41 V dans le couple Cr3+/Cr2+) existent en faux équilibre.Dans le cas d’oxydants ou de réducteurs extrêmement énergiques, les réactions sur l’eau sont rapides. C’est le cas, par exemple, du fluor (E0 = 2,87 V) ou du sodium (E0 = 漣 2,71 V) qui réagissent quantitativement sur l’eau selon: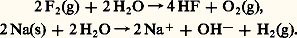 TitragesLa relation de Nernst montre que le potentiel d’oxydoréduction d’une solution est représentatif de l’état de la solution. Cette propriété est mise à profit dans les titrages. Les variations du potentiel sont suivies au cours de l’addition par exemple d’un oxydant dans une solution de réducteur. En pratique, ce sont des réactions très quantitatives qui sont choisies, par exemple l’oxydation du fer (II) par le cérium (IV). Le point équivalent se caractérise alors par une variation brutale du potentiel (fig. 6). Les variations du potentiel au voisinage de la demi-équivalence sont au contraire lentes. On a alors une solution-tampon de potentiel. Il existe aussi des indicateurs colorés d’oxydoréduction: ils changent de couleur dans une certaine plage de potentiel.3. Réactions de formation de complexes (transfert d’ions ou de molécules polaires)DéfinitionsComplexes. Couple donneur-accepteur d’une particuleUn complexe est une entité formée de l’association de deux ou plusieurs espèces chimiques, ions ou molécules polaires. Les complexes métalliques, qui réunissent un cation métallique et une ou plusieurs espèces «complexantes» avec échange de liaisons de coordination, sont les plus importants en pratique (cf. COMPLEXES [chimie]).Par exemple, l’ion FeSC2+ est un complexe formé entre les ions Fe3+ et SC-, Cu(NH3)42+ est un complexe formé entre l’ion Cu2+ et quatre molécules d’ammoniac (molécule polaire), FeY- un complexe formé entre le cation Fe3+ et l’ion Y4-, Y4- symbolisant l’anion éthylènediaminetétracétate (E.D.T.A.), réactif chélatant des cations métalliques d’usage courant.Chaque complexe peut donc être défini comme un donneur de particule, celle-ci pouvant être un ion ou une molécule polaire. L’autre espèce joue alors le rôle d’accepteur conjugué dans le couple:
TitragesLa relation de Nernst montre que le potentiel d’oxydoréduction d’une solution est représentatif de l’état de la solution. Cette propriété est mise à profit dans les titrages. Les variations du potentiel sont suivies au cours de l’addition par exemple d’un oxydant dans une solution de réducteur. En pratique, ce sont des réactions très quantitatives qui sont choisies, par exemple l’oxydation du fer (II) par le cérium (IV). Le point équivalent se caractérise alors par une variation brutale du potentiel (fig. 6). Les variations du potentiel au voisinage de la demi-équivalence sont au contraire lentes. On a alors une solution-tampon de potentiel. Il existe aussi des indicateurs colorés d’oxydoréduction: ils changent de couleur dans une certaine plage de potentiel.3. Réactions de formation de complexes (transfert d’ions ou de molécules polaires)DéfinitionsComplexes. Couple donneur-accepteur d’une particuleUn complexe est une entité formée de l’association de deux ou plusieurs espèces chimiques, ions ou molécules polaires. Les complexes métalliques, qui réunissent un cation métallique et une ou plusieurs espèces «complexantes» avec échange de liaisons de coordination, sont les plus importants en pratique (cf. COMPLEXES [chimie]).Par exemple, l’ion FeSC2+ est un complexe formé entre les ions Fe3+ et SC-, Cu(NH3)42+ est un complexe formé entre l’ion Cu2+ et quatre molécules d’ammoniac (molécule polaire), FeY- un complexe formé entre le cation Fe3+ et l’ion Y4-, Y4- symbolisant l’anion éthylènediaminetétracétate (E.D.T.A.), réactif chélatant des cations métalliques d’usage courant.Chaque complexe peut donc être défini comme un donneur de particule, celle-ci pouvant être un ion ou une molécule polaire. L’autre espèce joue alors le rôle d’accepteur conjugué dans le couple: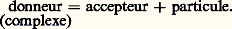 Par exemple, le complexe FeSC2+ peut être considéré comme un donneur de thiocyanate (SCN- est la particule, Fe3+ est l’accepteur) ou comme un donneur de Fe3+ (Fe3+ est la particule, SC- est l’accepteur). De même, d’après la définition précédente, Cu(NH3)42+ apparaît comme un polydonneur de la particule NH3.Les acides, qui sont des donneurs de la particule H+ dans le modèle de Brönsted introduit précédemment, peuvent être considérés comme des complexes. L’étude des réactions de formation de complexes concerne l’ensemble des particules autres que H+ et l’électron. L’introduction des couples donneur-accepteur d’une particule quelconque correspond à une généralisation des concepts d’acido-basicité. Celle-ci apparaît clairement dans la définition des acides et des bases proposée par Lewis, selon laquelle un acide, électrophile, est porteur d’une lacune électronique (orbitales vacantes facilement accessibles du point de vue énergétique), et une base, nucléophile, est un donneur de doublet électronique. L’établissement d’une liaison entre ces deux entités, appelée réaction acide-base dans ce nouveau modèle, correspond de fait à ce que nous appelons ici la formation d’un complexe.Réactions d’échange d’ions ou de molécules polairesEn règle générale, les particules n’existent pas à l’état libre en solution (elles n’y existent qu’à l’état solvaté), de telle sorte que pour qu’un donneur (donn1) cède une particule p , il faut qu’il se trouve en présence d’un accepteur (acc2) de la même particule. Entre les deux couples:
Par exemple, le complexe FeSC2+ peut être considéré comme un donneur de thiocyanate (SCN- est la particule, Fe3+ est l’accepteur) ou comme un donneur de Fe3+ (Fe3+ est la particule, SC- est l’accepteur). De même, d’après la définition précédente, Cu(NH3)42+ apparaît comme un polydonneur de la particule NH3.Les acides, qui sont des donneurs de la particule H+ dans le modèle de Brönsted introduit précédemment, peuvent être considérés comme des complexes. L’étude des réactions de formation de complexes concerne l’ensemble des particules autres que H+ et l’électron. L’introduction des couples donneur-accepteur d’une particule quelconque correspond à une généralisation des concepts d’acido-basicité. Celle-ci apparaît clairement dans la définition des acides et des bases proposée par Lewis, selon laquelle un acide, électrophile, est porteur d’une lacune électronique (orbitales vacantes facilement accessibles du point de vue énergétique), et une base, nucléophile, est un donneur de doublet électronique. L’établissement d’une liaison entre ces deux entités, appelée réaction acide-base dans ce nouveau modèle, correspond de fait à ce que nous appelons ici la formation d’un complexe.Réactions d’échange d’ions ou de molécules polairesEn règle générale, les particules n’existent pas à l’état libre en solution (elles n’y existent qu’à l’état solvaté), de telle sorte que pour qu’un donneur (donn1) cède une particule p , il faut qu’il se trouve en présence d’un accepteur (acc2) de la même particule. Entre les deux couples: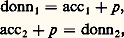 on a la réaction globale:
on a la réaction globale: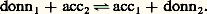 Celle-ci montre que, là encore, on ne peut envisager que des réactions de transfert entre le donneur d’un système et l’accepteur d’un autre système. La nécessité pour les deux couples de mettre en jeu la même particule permet de lever l’ambiguïté précédente quant au choix de son identité. Ainsi, la réaction entre FeSC2+ et - se traite en termes d’échange de la particule Fe3+:
Celle-ci montre que, là encore, on ne peut envisager que des réactions de transfert entre le donneur d’un système et l’accepteur d’un autre système. La nécessité pour les deux couples de mettre en jeu la même particule permet de lever l’ambiguïté précédente quant au choix de son identité. Ainsi, la réaction entre FeSC2+ et - se traite en termes d’échange de la particule Fe3+: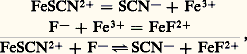 alors que la réaction entre MgY2- et Fe3+ correspond à un transfert de la particule Y4-:
alors que la réaction entre MgY2- et Fe3+ correspond à un transfert de la particule Y4-: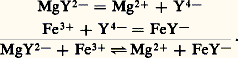 De nouveau, à l’écriture de ces réactions de transfert, apparaît l’intérêt d’établir un classement des donneurs (ou des accepteurs) d’une même particule, pour permettre de prévoir simplement le sens prédominant de telles réactions.révision des réactionsPouvoir accepteur du solvant. Constante de stabilitéLa dissolution dans un solvant résulte de la solvatation des espèces par celui-ci. Le solvant peut donc jouer le rôle d’accepteur de toute particule:
De nouveau, à l’écriture de ces réactions de transfert, apparaît l’intérêt d’établir un classement des donneurs (ou des accepteurs) d’une même particule, pour permettre de prévoir simplement le sens prédominant de telles réactions.révision des réactionsPouvoir accepteur du solvant. Constante de stabilitéLa dissolution dans un solvant résulte de la solvatation des espèces par celui-ci. Le solvant peut donc jouer le rôle d’accepteur de toute particule: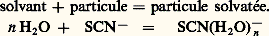 D’où l’idée d’établir le classement des donneurs d’une même particule d’après l’intensité de leur réaction sur le solvant qui joue le rôle d’accepteur de cette particule. Ainsi, l’écriture classique de la dissociation (hydrolyse) du complexe FeSC2+ en solution aqueuse:
D’où l’idée d’établir le classement des donneurs d’une même particule d’après l’intensité de leur réaction sur le solvant qui joue le rôle d’accepteur de cette particule. Ainsi, l’écriture classique de la dissociation (hydrolyse) du complexe FeSC2+ en solution aqueuse: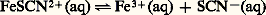 peut être considérée comme l’expression du transfert de la particule SC- du donneur FeSC2+ à l’accepteur H2O. Pour chaque complexe thiocyanate et partant d’une même concentration introduite, on aura affaire à un donneur de cette particule d’autant plus fort que la concentration de SC- à l’équilibre sera plus grande, soit encore pSCN = 漣 lg|SC-| petit.À l’équilibre précédent correspond la constante de dissociation:
peut être considérée comme l’expression du transfert de la particule SC- du donneur FeSC2+ à l’accepteur H2O. Pour chaque complexe thiocyanate et partant d’une même concentration introduite, on aura affaire à un donneur de cette particule d’autant plus fort que la concentration de SC- à l’équilibre sera plus grande, soit encore pSCN = 漣 lg|SC-| petit.À l’équilibre précédent correspond la constante de dissociation: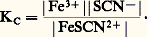 On voit que la donnée de KC (ou plus commodément pKC = 漣 lgKC) caractérise la force du donneur et de l’accepteur. Plus KC est grand, ou pKC petit, plus le donneur est fort (c’est-à-dire moins le complexe est stable) et réciproquement.Il faut noter l’analogie entre pSCN, ou plus généralement pX (X désignant une particule solvatée quelconque), et le pH d’une solution. De même qu’il existe des électrodes pour la mesure du pH, il existe des électrodes dites «sélectives», «spécifiques», ou encore «indicatrices de concentration d’ions ou de molécules», permettant la détermination de pX pour certaines espèces en solution.Certains auteurs caractérisent la stabilité des complexes par la constante de formation KF (notée aussi 廓), qui est l’inverse de la constante de dissociation:
On voit que la donnée de KC (ou plus commodément pKC = 漣 lgKC) caractérise la force du donneur et de l’accepteur. Plus KC est grand, ou pKC petit, plus le donneur est fort (c’est-à-dire moins le complexe est stable) et réciproquement.Il faut noter l’analogie entre pSCN, ou plus généralement pX (X désignant une particule solvatée quelconque), et le pH d’une solution. De même qu’il existe des électrodes pour la mesure du pH, il existe des électrodes dites «sélectives», «spécifiques», ou encore «indicatrices de concentration d’ions ou de molécules», permettant la détermination de pX pour certaines espèces en solution.Certains auteurs caractérisent la stabilité des complexes par la constante de formation KF (notée aussi 廓), qui est l’inverse de la constante de dissociation: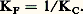 Les électrolytes forts, entièrement dissociés en leurs ions en solution, sont des donneurs forts (réaction sur le solvant quantitative). Lorsque le solvant est lui-même donneur de la particule (par exemple NH2- dans l’ammoniaque liquide, C1- ou 2- dans certains sels fondus), l’étude des complexes correspondants conduit à l’introduction de la solvoacidité (par analogie avec les réactions d’échange du H+ dans HS).Échelle de pXConsidérons la réaction d’échange:
Les électrolytes forts, entièrement dissociés en leurs ions en solution, sont des donneurs forts (réaction sur le solvant quantitative). Lorsque le solvant est lui-même donneur de la particule (par exemple NH2- dans l’ammoniaque liquide, C1- ou 2- dans certains sels fondus), l’étude des complexes correspondants conduit à l’introduction de la solvoacidité (par analogie avec les réactions d’échange du H+ dans HS).Échelle de pXConsidérons la réaction d’échange: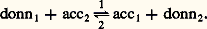 La constante Kéq de cet équilibre est donnée par:
La constante Kéq de cet équilibre est donnée par: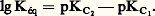 Le sens prépondérant de la réaction de transfert est le sens 1 ou le sens 2 selon que la constante Kéq est grande ou, au contraire, petite devant 1, d’où la règle: pour que le donneur d’un système réagisse avec l’accepteur d’un autre système mettant en jeu la même particule, il faut que le pKC du premier couple soit inférieur à celui du second, et la réaction est d’autant plus quantitative que l’écart est plus grand.Il est commode, pour effectuer des prévisions, d’utiliser une échelle de pX(= 漣 lg|X|, |X| désignant la concentration de la particule solvatée), sur laquelle on repère chaque couple donneur-accepteur de la particule par la valeur de pKC, valeur de pX pour une solution contenant, à l’équilibre, des concentrations égales des deux formes conjuguées.Les figures 7 a et 7 b montrent, à titre d’exemple, une échelle de pY = 漣 lg|Y4-| permettant la prévision des réactions en solution aqueuse entre l’E.D.T.A. et un cation métallique, et une échelle de pFe = 漣 lg|Fe3+| pour la prévision des réactions entre le fer (III) et les complexants de celui-ci. La place des différents couples sur l’échelle de pY montre que le complexe MgY2- réagit quantitativement avec Fe3+, pour former un complexe beaucoup plus stable:
Le sens prépondérant de la réaction de transfert est le sens 1 ou le sens 2 selon que la constante Kéq est grande ou, au contraire, petite devant 1, d’où la règle: pour que le donneur d’un système réagisse avec l’accepteur d’un autre système mettant en jeu la même particule, il faut que le pKC du premier couple soit inférieur à celui du second, et la réaction est d’autant plus quantitative que l’écart est plus grand.Il est commode, pour effectuer des prévisions, d’utiliser une échelle de pX(= 漣 lg|X|, |X| désignant la concentration de la particule solvatée), sur laquelle on repère chaque couple donneur-accepteur de la particule par la valeur de pKC, valeur de pX pour une solution contenant, à l’équilibre, des concentrations égales des deux formes conjuguées.Les figures 7 a et 7 b montrent, à titre d’exemple, une échelle de pY = 漣 lg|Y4-| permettant la prévision des réactions en solution aqueuse entre l’E.D.T.A. et un cation métallique, et une échelle de pFe = 漣 lg|Fe3+| pour la prévision des réactions entre le fer (III) et les complexants de celui-ci. La place des différents couples sur l’échelle de pY montre que le complexe MgY2- réagit quantitativement avec Fe3+, pour former un complexe beaucoup plus stable: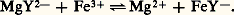 On lit directement sur l’échelle:
On lit directement sur l’échelle: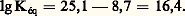 De même, considérant la seconde échelle, on explique aisément pourquoi FeSC2+, complexe rouge, est détruit par addition de fluorure, pour former Fe2+, complexe incolore, plus stable:
De même, considérant la seconde échelle, on explique aisément pourquoi FeSC2+, complexe rouge, est détruit par addition de fluorure, pour former Fe2+, complexe incolore, plus stable: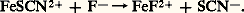 L’addition de complexants auxiliaires, donnant des complexes très stables avec certaines espèces, permet de «dissimuler» celles-ci vis-à-vis de certains de leurs réactifs (ceux, par exemple, qui produisent des complexes moins stables); cet effet, connu sous le nom de masquage , est largement utilisé, lors de titrages complexométriques, pour leur conférer un caractère sélectif.Complexes successifs. Domaines de prédominanceLes cations métalliques de coordinence élevée peuvent fixer successivement plusieurs ligands. Ainsi, l’addition d’oxalate de sodium (donneur fort de la particule C242-) à une solution aqueuse de fer ferrique conduit à la formation, dans l’ordre, des complexes Fe(C24)+, Fe(C24)2-, Fe(C24)33-.La figure 8 montre les variations simulées du pFe, pC24 et des pourcentages des différentes formes du fer en solution au cours d’un tel titrage.4. Réactions de précipitationUn grand nombre de composés ont une solubilité limitée. Citons la plupart des sulfures, des orthophosphates et des hydroxydes, les oxalates et les sulfates alcalino-terreux, les halogénures d’argent, les oxinates...Produit de solubilité et solubilitéProduit de solubilitéSoit la réaction de formation de complexe:
L’addition de complexants auxiliaires, donnant des complexes très stables avec certaines espèces, permet de «dissimuler» celles-ci vis-à-vis de certains de leurs réactifs (ceux, par exemple, qui produisent des complexes moins stables); cet effet, connu sous le nom de masquage , est largement utilisé, lors de titrages complexométriques, pour leur conférer un caractère sélectif.Complexes successifs. Domaines de prédominanceLes cations métalliques de coordinence élevée peuvent fixer successivement plusieurs ligands. Ainsi, l’addition d’oxalate de sodium (donneur fort de la particule C242-) à une solution aqueuse de fer ferrique conduit à la formation, dans l’ordre, des complexes Fe(C24)+, Fe(C24)2-, Fe(C24)33-.La figure 8 montre les variations simulées du pFe, pC24 et des pourcentages des différentes formes du fer en solution au cours d’un tel titrage.4. Réactions de précipitationUn grand nombre de composés ont une solubilité limitée. Citons la plupart des sulfures, des orthophosphates et des hydroxydes, les oxalates et les sulfates alcalino-terreux, les halogénures d’argent, les oxinates...Produit de solubilité et solubilitéProduit de solubilitéSoit la réaction de formation de complexe: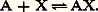 Supposons que AX soit peu soluble. Deux situations peuvent se présenter.1. La solution est saturée de AX; du solide est alors en excès et on a les deux équilibres simultanés:
Supposons que AX soit peu soluble. Deux situations peuvent se présenter.1. La solution est saturée de AX; du solide est alors en excès et on a les deux équilibres simultanés: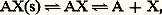 (s) désignant la phase solide.Par exemple:
(s) désignant la phase solide.Par exemple: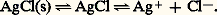 L’application de la loi d’action de masses aux deux équilibres donne:– pour le premier |AX| = S0 (S0 étant la solubilité propre de AX);– pour le second:
L’application de la loi d’action de masses aux deux équilibres donne:– pour le premier |AX| = S0 (S0 étant la solubilité propre de AX);– pour le second: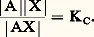 Des deux relations précédentes on tire:
Des deux relations précédentes on tire: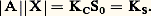 KS est appelé produit de solubilité de AX. On utilise plus commodément pKS = 漣 lgKS. Dans le cas du chlorure d’argent:
KS est appelé produit de solubilité de AX. On utilise plus commodément pKS = 漣 lgKS. Dans le cas du chlorure d’argent: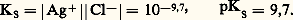 2. La solution n’est pas saturée de AX; dans ce cas |AX| 麗 S0 et, par conséquent:
2. La solution n’est pas saturée de AX; dans ce cas |AX| 麗 S0 et, par conséquent: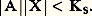 Jamais on ne pourra avoir |A||X| 礪 KS. Si, dans une solution, les concentrations |A| et |X| introduites semblaient devoir vérifier cette dernière inégalité, il précipiterait AX en quantité telle que le produit des concentrations résiduelles |A| et |X| vérifieraient l’égalité au produit de solubilité.Dans le cas général:
Jamais on ne pourra avoir |A||X| 礪 KS. Si, dans une solution, les concentrations |A| et |X| introduites semblaient devoir vérifier cette dernière inégalité, il précipiterait AX en quantité telle que le produit des concentrations résiduelles |A| et |X| vérifieraient l’égalité au produit de solubilité.Dans le cas général: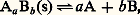 la relation du produit de solubilité s’écrit:
la relation du produit de solubilité s’écrit: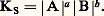 Par exemple, pour Ag2Cr4:
Par exemple, pour Ag2Cr4: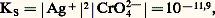 pour Ca3(PO4)2,
pour Ca3(PO4)2,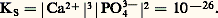 Certains composés sont extrêmement insolubles, comme le sulfure mercurique HgS avec |Hg2+||S2-| = 10-51,8.Solubilité totaleLa solubilité totale S d’un composé AX (exprimée en moles de A dissoutes) est la somme des concentrations de A sous toutes ses formes quand on introduit AX jusqu’à saturation dans une solution quelconque:
Certains composés sont extrêmement insolubles, comme le sulfure mercurique HgS avec |Hg2+||S2-| = 10-51,8.Solubilité totaleLa solubilité totale S d’un composé AX (exprimée en moles de A dissoutes) est la somme des concentrations de A sous toutes ses formes quand on introduit AX jusqu’à saturation dans une solution quelconque: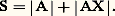 Or |AX| = S0 et |A||X| =KS; par suite:
Or |AX| = S0 et |A||X| =KS; par suite: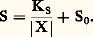 Elle dépend de la présence initiale éventuelle de X. On pourrait, de même, exprimer la solubilité totale de AX en moles de X dissoutes.Solubilité dans l’eau pureSi on dissout AX dans l’eau pure, on a:
Elle dépend de la présence initiale éventuelle de X. On pourrait, de même, exprimer la solubilité totale de AX en moles de X dissoutes.Solubilité dans l’eau pureSi on dissout AX dans l’eau pure, on a: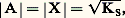 et la relation du paragraphe précédent s’écrit:
et la relation du paragraphe précédent s’écrit: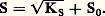 Le terme S0 est souvent très faible et négligeable. On obtient alors:
Le terme S0 est souvent très faible et négligeable. On obtient alors: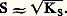 Cette approximation est justifiée dans le cas du chlorure d’argent AgCl. En effet, S0 = 2 憐 10-7 mole . l-1 et KS = |Ag+||Cl-| = 2 憐 10-10, d’où:
Cette approximation est justifiée dans le cas du chlorure d’argent AgCl. En effet, S0 = 2 憐 10-7 mole . l-1 et KS = |Ag+||Cl-| = 2 憐 10-10, d’où: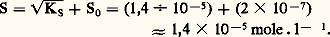 Dans quelques cas, elle ne l’est pas. C’est ainsi que pour l’acétate d’argent AgCH3COO:
Dans quelques cas, elle ne l’est pas. C’est ainsi que pour l’acétate d’argent AgCH3COO: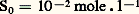 et
et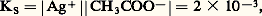 d’où:
d’où: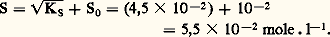 S0 représente près de 20 p. 100 de la solubilité totale de l’acétate d’argent.Pour un composé de stœchiométrie différente, par exemple le chromate d’argent Ag2Cr4, le produit de solubilité s’écrit:
S0 représente près de 20 p. 100 de la solubilité totale de l’acétate d’argent.Pour un composé de stœchiométrie différente, par exemple le chromate d’argent Ag2Cr4, le produit de solubilité s’écrit: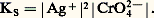 Si on pose |Ag2Cr4| = S0, la solubilité totale peut s’écrire de deux manières selon qu’on l’exprime en moles de Ag+ ou en moles de Cr42-, soit respectivement:
Si on pose |Ag2Cr4| = S0, la solubilité totale peut s’écrire de deux manières selon qu’on l’exprime en moles de Ag+ ou en moles de Cr42-, soit respectivement: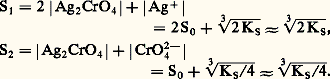 Précipitation par un excès de réactifConsidérons une solution d’ions argent Ag+ à 0,1 mole . l-1 dans laquelle on ajoute des ions chlorure Cl-. Le chlorure d’argent précipite dès que le produit de solubilité est atteint, c’est-à-dire dès que:
Précipitation par un excès de réactifConsidérons une solution d’ions argent Ag+ à 0,1 mole . l-1 dans laquelle on ajoute des ions chlorure Cl-. Le chlorure d’argent précipite dès que le produit de solubilité est atteint, c’est-à-dire dès que: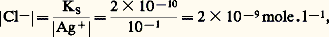 en pratique dès la première goutte.S0 étant négligeable et sachant qu’il n’existe pas de complexe supérieur, la solubilité du chlorure d’argent s’écrit:
en pratique dès la première goutte.S0 étant négligeable et sachant qu’il n’existe pas de complexe supérieur, la solubilité du chlorure d’argent s’écrit: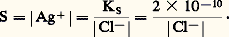 Elle dépend de la concentration: plus |Cl-| est élevée, plus la précipitation est quantitative. Une précipitation complète de l’argent à mieux que 0,1 p.100 près nécessite, outre la quantité de chlorure entraînée dans le précipité, une quantité telle que:
Elle dépend de la concentration: plus |Cl-| est élevée, plus la précipitation est quantitative. Une précipitation complète de l’argent à mieux que 0,1 p.100 près nécessite, outre la quantité de chlorure entraînée dans le précipité, une quantité telle que: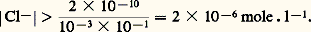 Si on dispose d’un moyen (électrode sélective par exemple) permettant de suivre les variations de concentration de l’une des deux espèces réagissantes, la précipitation par ajout d’un réactif devient une méthode de titrage. Dans l’exemple considéré, un fil d’argent, par l’intermédiaire de son potentiel (E = 0,80 V + 0,06lg|Ag+|), est indicateur de pAg = 漣 lg|Ag+|. On obtient la courbe de la figure 9; le passage par le point équivalent se traduit par une variation brutale du potentiel (ou de pAg).Cinétique des réactions de précipitationLa formation et l’évolution des précipités vers un équilibre stable sont rarement des processus instantanés. La précipitation commence par la formation de germes cristallins constitués par l’association de quelques ions. Ces germes grossissent par fixation (adsorption) de nouveaux ions. Si la formation des germes est lente, il y a sursaturation. Elle peut être accélérée par agitation, par introduction de cristaux déjà formés. La concentration et la nature même du sel interviennent aussi.La surface et même l’intérieur des cristaux sont le siège d’un échange incessant avec les ions de la solution. Peu à peu, le cristal s’organise et se perfectionne, sa taille augmente dans le temps et sa solubilité diminue. La solubilité de l’hydroxyde de magnésium Mg(OH)2 peut passer ainsi de 10-3 mole . l-1 à 10-4 mole . l-1 en vingt-quatre heures.La plupart des hydroxydes sont insolubles; ils sont rarement purs en raison des phénomènes d’adsorption. Certains se déshydratent spontanément par ébullition (AgOH, Hg(OH)2 ...).Solubilité et aciditéLa solubilité des composés peu solubles varie avec le pH de la solution si l’un des constituants du précipité possède des propriétés acides ou basiques. C’est un cas extrêmement fréquent: citons les hydroxydes, les sulfures, les oxalates, les oxinates... Cette propriété a été largement utilisée en analyse qualitative ou quantitative pour la précipitation sélective des cations métalliques (séparation des cations alcalino-terreux par précipitation des oxalates par exemple).Solubilité des hydroxydesConsidérons le cas de l’aluminium. Son hydroxyde Al(OH)3 est une base selon l’équilibre:
Si on dispose d’un moyen (électrode sélective par exemple) permettant de suivre les variations de concentration de l’une des deux espèces réagissantes, la précipitation par ajout d’un réactif devient une méthode de titrage. Dans l’exemple considéré, un fil d’argent, par l’intermédiaire de son potentiel (E = 0,80 V + 0,06lg|Ag+|), est indicateur de pAg = 漣 lg|Ag+|. On obtient la courbe de la figure 9; le passage par le point équivalent se traduit par une variation brutale du potentiel (ou de pAg).Cinétique des réactions de précipitationLa formation et l’évolution des précipités vers un équilibre stable sont rarement des processus instantanés. La précipitation commence par la formation de germes cristallins constitués par l’association de quelques ions. Ces germes grossissent par fixation (adsorption) de nouveaux ions. Si la formation des germes est lente, il y a sursaturation. Elle peut être accélérée par agitation, par introduction de cristaux déjà formés. La concentration et la nature même du sel interviennent aussi.La surface et même l’intérieur des cristaux sont le siège d’un échange incessant avec les ions de la solution. Peu à peu, le cristal s’organise et se perfectionne, sa taille augmente dans le temps et sa solubilité diminue. La solubilité de l’hydroxyde de magnésium Mg(OH)2 peut passer ainsi de 10-3 mole . l-1 à 10-4 mole . l-1 en vingt-quatre heures.La plupart des hydroxydes sont insolubles; ils sont rarement purs en raison des phénomènes d’adsorption. Certains se déshydratent spontanément par ébullition (AgOH, Hg(OH)2 ...).Solubilité et aciditéLa solubilité des composés peu solubles varie avec le pH de la solution si l’un des constituants du précipité possède des propriétés acides ou basiques. C’est un cas extrêmement fréquent: citons les hydroxydes, les sulfures, les oxalates, les oxinates... Cette propriété a été largement utilisée en analyse qualitative ou quantitative pour la précipitation sélective des cations métalliques (séparation des cations alcalino-terreux par précipitation des oxalates par exemple).Solubilité des hydroxydesConsidérons le cas de l’aluminium. Son hydroxyde Al(OH)3 est une base selon l’équilibre: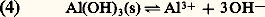 avec:
avec: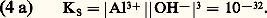 C’est aussi un acide selon l’équilibre:
C’est aussi un acide selon l’équilibre: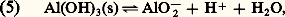 avec:
avec: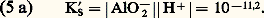 Les règles classiques du déplacement des équilibres chimiques montrent qu’en milieu acide l’équilibre (4) est déplacé vers la droite: l’hydroxyde se redissout sous forme Al3+. En milieu basique, l’équilibre (5) est déplacé vers la droite: l’hydroxyde se redissout sous forme Al2-. Si on admet que la solubilité propre S0 de Al(OH)3 est négligeable, la solubilité totale s’exprime par:
Les règles classiques du déplacement des équilibres chimiques montrent qu’en milieu acide l’équilibre (4) est déplacé vers la droite: l’hydroxyde se redissout sous forme Al3+. En milieu basique, l’équilibre (5) est déplacé vers la droite: l’hydroxyde se redissout sous forme Al2-. Si on admet que la solubilité propre S0 de Al(OH)3 est négligeable, la solubilité totale s’exprime par: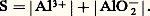 En présence de précipité, les produits de solubilité KS et K S sont vérifiés et S s’écrit:
En présence de précipité, les produits de solubilité KS et K S sont vérifiés et S s’écrit: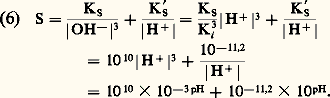 On peut tracer le diagramme représentatif des variations de solubilité en fonction du pH. On l’appelle «diagramme de solubilité» (fig. 10). Il peut aussi être mis sous forme logarithmique, ce qui visualise mieux la valeur du minimum de solubilité (face=F0019 漣 lgS est porté en fonction du pH). L’expression (6) n’est valable qu’en présence du précipité, donc dans un domaine de pH qui dépend de la concentration. Si on choisit une concentration totale en aluminium de 10-2 mole . l-1, d’après (4 a) et (5 a), l’expression (6) n’est valable qu’entre pH = 4 (début de précipitation de l’hydroxyde) et pH = 9,2 (fin de redissolution ou début de précipitation quand on vient des milieux basiques).On peut tracer de tels diagrammes de solubilité pour les hydroxydes des autres éléments (fig. 11). Ils ont toujours la même allure. Toutefois, dans le domaine de pH usuel (0-14), la redissolution vers les pH les plus élevés n’est observée que dans le cas de l’aluminium, du chrome, du zinc, du plomb.5. Équilibres variésIl est assez rare que les types de réactions évoqués précédemment (acido-basicité, oxydoréduction, formation de complexes, précipitation) se produisent seuls. Il a déjà été fait allusion à l’influence de l’acidité sur les propriétés oxydoréductrices et sur la solubilité. D’autres associations de deux de ces phénomènes présentent un grand intérêt.Oxydoréduction et complexes. Oxydoréduction et précipitationOn peut modifier les propriétés oxydoréductrices par formation de complexes. Considérons le couple:
On peut tracer le diagramme représentatif des variations de solubilité en fonction du pH. On l’appelle «diagramme de solubilité» (fig. 10). Il peut aussi être mis sous forme logarithmique, ce qui visualise mieux la valeur du minimum de solubilité (face=F0019 漣 lgS est porté en fonction du pH). L’expression (6) n’est valable qu’en présence du précipité, donc dans un domaine de pH qui dépend de la concentration. Si on choisit une concentration totale en aluminium de 10-2 mole . l-1, d’après (4 a) et (5 a), l’expression (6) n’est valable qu’entre pH = 4 (début de précipitation de l’hydroxyde) et pH = 9,2 (fin de redissolution ou début de précipitation quand on vient des milieux basiques).On peut tracer de tels diagrammes de solubilité pour les hydroxydes des autres éléments (fig. 11). Ils ont toujours la même allure. Toutefois, dans le domaine de pH usuel (0-14), la redissolution vers les pH les plus élevés n’est observée que dans le cas de l’aluminium, du chrome, du zinc, du plomb.5. Équilibres variésIl est assez rare que les types de réactions évoqués précédemment (acido-basicité, oxydoréduction, formation de complexes, précipitation) se produisent seuls. Il a déjà été fait allusion à l’influence de l’acidité sur les propriétés oxydoréductrices et sur la solubilité. D’autres associations de deux de ces phénomènes présentent un grand intérêt.Oxydoréduction et complexes. Oxydoréduction et précipitationOn peut modifier les propriétés oxydoréductrices par formation de complexes. Considérons le couple: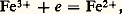 avec:
avec: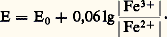 En milieu fluorure, Fe3+ est complexé selon:
En milieu fluorure, Fe3+ est complexé selon: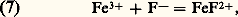 avec:
avec: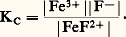 Si, à une solution de Fe3+ et de Fe2+ on ajoute -, |Fe3+| diminue et E également. Les ions ferriques sont devenus moins oxydants en présence de fluorure, ou encore les ions ferreux sont devenus plus réducteurs. Tout se passe comme si E0 avait diminué. En milieu fluorure le couple redox s’écrit:
Si, à une solution de Fe3+ et de Fe2+ on ajoute -, |Fe3+| diminue et E également. Les ions ferriques sont devenus moins oxydants en présence de fluorure, ou encore les ions ferreux sont devenus plus réducteurs. Tout se passe comme si E0 avait diminué. En milieu fluorure le couple redox s’écrit: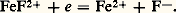 Si on applique la loi de Nernst:
Si on applique la loi de Nernst: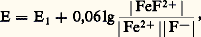 on peut facilement montrer que:
on peut facilement montrer que: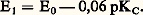 E1 est le potentiel normal apparent en milieu fluorure.Dans le cas plus général où l’oxydant et le réducteur sont complexés, on démontre:
E1 est le potentiel normal apparent en milieu fluorure.Dans le cas plus général où l’oxydant et le réducteur sont complexés, on démontre: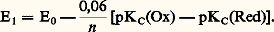 De même, la précipitation d’un oxydant ou de son réducteur conjugué modifie les propriétés du couple; ainsi, en milieu chlorure, le couple Ag+/Ag devient AgCl(s) + e = Ag(s) + Cl- avec KS = |Ag+||Cl-|. Le potentiel normal E0 doit être remplacé par le potentiel normal apparent:
De même, la précipitation d’un oxydant ou de son réducteur conjugué modifie les propriétés du couple; ainsi, en milieu chlorure, le couple Ag+/Ag devient AgCl(s) + e = Ag(s) + Cl- avec KS = |Ag+||Cl-|. Le potentiel normal E0 doit être remplacé par le potentiel normal apparent: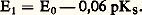 Cette modification des propriétés redox est très importante en pratique. Ainsi, l’or est très difficile à oxyder. Le couple Au3+/Au(s) a un potentiel normal E0 = 1,50 V et Au+ est instable (il se dismute en Au3+ et Au(s)). En milieu cyanure, Au(I) existe sous forme de complexe cyanure et est stabilisé. L’oxydation de l’or en Au(I) devient possible par l’oxygène (couple Au(CN)2-/Au(s), E 0 = 0,06 V). Cette réaction est mise en œuvre industriellement pour le traitement des minerais d’or.Complexes et aciditéOn peut modifier la stabilité d’un complexe en jouant sur le pH, ou inversement, modifier la force d’un acide ou d’une base par formation de complexes. Considérons l’équilibre (7) et l’équilibre H+ + - = HF, qui montre que - est une base. En milieu acide les ions fluorure sont consommés pour former HF et l’équilibre (7) se déplace vers la gauche: le complexe FeF2+ est détruit. On peut écrire l’équilibre global:
Cette modification des propriétés redox est très importante en pratique. Ainsi, l’or est très difficile à oxyder. Le couple Au3+/Au(s) a un potentiel normal E0 = 1,50 V et Au+ est instable (il se dismute en Au3+ et Au(s)). En milieu cyanure, Au(I) existe sous forme de complexe cyanure et est stabilisé. L’oxydation de l’or en Au(I) devient possible par l’oxygène (couple Au(CN)2-/Au(s), E 0 = 0,06 V). Cette réaction est mise en œuvre industriellement pour le traitement des minerais d’or.Complexes et aciditéOn peut modifier la stabilité d’un complexe en jouant sur le pH, ou inversement, modifier la force d’un acide ou d’une base par formation de complexes. Considérons l’équilibre (7) et l’équilibre H+ + - = HF, qui montre que - est une base. En milieu acide les ions fluorure sont consommés pour former HF et l’équilibre (7) se déplace vers la gauche: le complexe FeF2+ est détruit. On peut écrire l’équilibre global: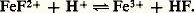 Il montre inversement que, en présence de Fe3+, l’acide fluorhydrique voit son acidité renforcée. Cette propriété peut être mise à profit pour renforcer l’acidité d’acides trop faibles pour être titrés avec précision en solution aqueuse. On peut citer, par exemple, le titrage de l’acide borique en présence de mannitol (fig. 12).Solubilité et formation de complexesOn peut détruire un complexe par précipitation ou, inversement, redissoudre un précipité par formation de complexes.Les ions argent Ag+ peuvent former, en présence d’ammoniac, le complexe argentodiammine Ag(NH3)2+ selon:
Il montre inversement que, en présence de Fe3+, l’acide fluorhydrique voit son acidité renforcée. Cette propriété peut être mise à profit pour renforcer l’acidité d’acides trop faibles pour être titrés avec précision en solution aqueuse. On peut citer, par exemple, le titrage de l’acide borique en présence de mannitol (fig. 12).Solubilité et formation de complexesOn peut détruire un complexe par précipitation ou, inversement, redissoudre un précipité par formation de complexes.Les ions argent Ag+ peuvent former, en présence d’ammoniac, le complexe argentodiammine Ag(NH3)2+ selon: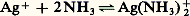 avec
avec Ils peuvent également précipiter en présence de chlorure:
Ils peuvent également précipiter en présence de chlorure: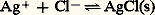 avec
avec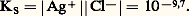 L’équilibre global s’écrit:
L’équilibre global s’écrit: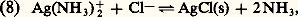 dont la constante d’équilibre vaut:
dont la constante d’équilibre vaut: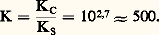 Cette valeur indique une tendance pour l’équilibre (8) à se déplacer vers la droite, c’est-à-dire que l’addition de chlorure dans une solution d’ions argentodiammine provoque la destruction de ce dernier avec précipitation de chlorure d’argent. Mais inversement, on peut redissoudre AgCl dans un excès d’ammoniaque. Le calcul montre qu’il est possible de dissoudre 10-2 mole de chlorure d’argent solide dans un litre d’ammoniaque à 0,22 mole . l-1.Il arrive que la redissolution d’un précipité puisse s’effectuer dans un excès du réactif précipitant. C’est ainsi que le cyanure d’argent peut se redissoudre à la faveur de la formation de complexes supérieurs:
Cette valeur indique une tendance pour l’équilibre (8) à se déplacer vers la droite, c’est-à-dire que l’addition de chlorure dans une solution d’ions argentodiammine provoque la destruction de ce dernier avec précipitation de chlorure d’argent. Mais inversement, on peut redissoudre AgCl dans un excès d’ammoniaque. Le calcul montre qu’il est possible de dissoudre 10-2 mole de chlorure d’argent solide dans un litre d’ammoniaque à 0,22 mole . l-1.Il arrive que la redissolution d’un précipité puisse s’effectuer dans un excès du réactif précipitant. C’est ainsi que le cyanure d’argent peut se redissoudre à la faveur de la formation de complexes supérieurs: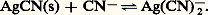 Les trois types de réactions en solution homogène se ramènent en fait à un type unique si on introduit la notion très générale de couple donneur-accepteur de particule. Le mot particule désigne toute entité échangeable en solution selon: donneur = accepteur + particule.Selon la nature de la particule, on retrouve les trois types évoqués (tabl. 4).Tous les raisonnements, tous les traitements mathématiques qui accompagnent ces trois types de réactions sont identiques sur le plan formel, à quelques nuances près, qui ont trait à l’intervention du solvant. Le solvant, en effet, n’est pas un milieu inerte mais un partenaire actif vis-à-vis de la particule échangée.L’eau en particulier est:– un acide et une base et, à ce titre, elle limite la force des acides et des bases qu’on y dissout;– un réducteur et un oxydant; elle devrait donc limiter la force des oxydants et des réducteurs, mais cette propriété ne se manifeste pratiquement pas pour des raisons cinétiques;– un accepteur d’ions ou de molécules polaires, et elle limite donc la force des donneurs seulement.L’eau n’est donneur que des ions qu’elle possède, c’est-à-dire H+ et OH-, et on retrouve alors l’acido-basicité.Le tableau 2 rassemble les caractéristiques des réactions de transfert d’une particule dans le cadre d’une présentation unitaire. Les principales relations quantitatives, dont on peut trouver les démonstrations et les domaines de validité dans les ouvrages spécialisés cités dans la bibliographie, y sont indiquées.
Les trois types de réactions en solution homogène se ramènent en fait à un type unique si on introduit la notion très générale de couple donneur-accepteur de particule. Le mot particule désigne toute entité échangeable en solution selon: donneur = accepteur + particule.Selon la nature de la particule, on retrouve les trois types évoqués (tabl. 4).Tous les raisonnements, tous les traitements mathématiques qui accompagnent ces trois types de réactions sont identiques sur le plan formel, à quelques nuances près, qui ont trait à l’intervention du solvant. Le solvant, en effet, n’est pas un milieu inerte mais un partenaire actif vis-à-vis de la particule échangée.L’eau en particulier est:– un acide et une base et, à ce titre, elle limite la force des acides et des bases qu’on y dissout;– un réducteur et un oxydant; elle devrait donc limiter la force des oxydants et des réducteurs, mais cette propriété ne se manifeste pratiquement pas pour des raisons cinétiques;– un accepteur d’ions ou de molécules polaires, et elle limite donc la force des donneurs seulement.L’eau n’est donneur que des ions qu’elle possède, c’est-à-dire H+ et OH-, et on retrouve alors l’acido-basicité.Le tableau 2 rassemble les caractéristiques des réactions de transfert d’une particule dans le cadre d’une présentation unitaire. Les principales relations quantitatives, dont on peut trouver les démonstrations et les domaines de validité dans les ouvrages spécialisés cités dans la bibliographie, y sont indiquées.
Encyclopédie Universelle. 2012.